

Abstract
Objective. Systemic sclerosis (SSc) is a rare connective tissue disease in childhood. We compared the characteristics of adult patients with juvenile-onset SSc (jSSc) from a single-center cohort to an adult-onset group.
Methods. Patients with disease onset before the age of 17 years were included in the jSSc cohort, while subjects with SSc onset after age 17 formed the adult-onset cohort.
Results. We identified 52 adult subjects with jSSc and compared them to 954 patients with adult-onset SSc. The mean ± SD age at disease onset of the patients with jSSc was 14 ± 2 years, 39 (75%) of them were women, and 24 (46%) had the diffuse cutaneous subset of SSc (dcSSc). There were no differences between the 2 cohorts in terms of sex and disease subset. Overlaps were significantly more frequent among the jSSc cohort (37%) compared to the adult-onset group (18%; p = 0.002). Autoantibody analysis demonstrated significantly more antitopoisomerase I antibody-positive subjects (33% vs 20%; p = 0.034) and significantly fewer anticentromere antibody-positive subjects (2% vs 25%; p < 0.001) in the jSSc cohort. Compared to the adult-onset group at 10 years from disease onset, survival was significantly higher among the subjects with jSSc (98% vs 75%; p = 0.001), pulmonary arterial hypertension had a significantly lower incidence (2% vs 14%; p = 0.032), and there was no difference in terms of pulmonary fibrosis (22% vs 21%) and cardiac scleroderma (3% vs 2%) between the 2 groups.
Conclusion. The high survival rates and lower proportion of dcSSc in the adult jSSc cohort may represent a survival bias.
Systemic sclerosis (SSc) is a rare autoimmune connective tissue disease in childhood. Annual incidence varies between 0.05 and 2.7 per 100,0001,2 and it has been reported that between 1.5% and 11.5% of all SSc cases develop in childhood3. Juvenile-onset SSc (jSSc) differs from adult-onset disease in both presentation and outcome. Over the last 10 years several large cohort studies assessing patients with jSSc were published, and comparison with adult-onset cohorts was made in 3 of them3,4,5. The study from the Pittsburgh Scleroderma Databank showed significantly higher frequency of overlap syndromes among the patients with jSSc with higher frequency of U1 ribonucleoprotein (RNP) antibodies, while anticentromere antibodies (ACA) and anti-RNA polymerase antibodies (ARA) were significantly underrepresented. The authors found no differences between childhood- and adult-onset disease in terms of lung and heart complications, while renal involvement was much more rare in the jSSc group. Interestingly, while the Pittsburgh jSSc group, which was derived from an adult SSc cohort, had subset distribution to the adult-onset group (dcSSc was found in 35% of childhood-onset patients), the diffuse subset was significantly overrepresented among the Paediatric Rheumatology European Society (PRES) jSSc cohort (91%)3,4. In contrast, the study using the EULAR (European League Against Rheumatism) Scleroderma Trial and Research (EUSTAR) cohort concentrated on those patients with jSSc who survived into adulthood, and found very few differences compared to adult-onset SSc5. The subset distribution, proportion of patients with overlap syndromes, and pattern of internal organ complications were very similar and the only significant difference was in the frequency of ACA, which was found in 5% of the juvenile-onset and 27% of the adult-onset group.
We investigated the characteristics of adult patients with childhood-onset SSc seen in our center and compared them to our adult-onset cohort.
MATERIALS AND METHODS
The Royal Free Hospital SSc database was filtered for adult subjects with childhood-onset disease (onset before the age of 17 years). Subjects were included according to the Paediatric Rheumatology European Society/American College of Rheumatology (ACR)/EULAR Provisional Classification Criteria for Juvenile Systemic Sclerosis6. All patients were secondary and tertiary referrals and the majority of them had been followed up under shared care. As a result, patients were reviewed in our center once every 6 to 18 months, while more frequent reviews were provided by their local specialists. To look for differences in disease course and outcome between childhood- and adult-onset disease, the jSSc cohort was compared to a large group of patients with adult-onset SSc also seen in our center. All subjects from the adult-onset cohort fulfilled the ACR preliminary classification criteria for SSc7.
Demographic and clinical characteristics were recorded for all patients and the data were collated and verified through patient case notes review. All subjects seen in our center have laboratory tests, including autoantibody profile, performed at the first visit, and for the majority, these are repeated usually once every 12 to 18 months. We used strict criteria to define internal organ complications, reflecting moderate to severe organ disease. Pulmonary fibrosis (PF) had to be confirmed on high-resolution computed tomography with either predicted forced vital capacity (FVC) < 55% or a 15% decline from baseline in FVC; or predicted carbon monoxide diffusing capacity (DLCO) < 55% or a 15% decline from baseline in DLCO, if pulmonary arterial hypertension (PAH) had been excluded. PAH had to be confirmed by right heart catheterization with mean pulmonary arterial pressure > 25 mm Hg and normal pulmonary capillary wedge pressure. Cardiac scleroderma was defined as hemodynamically significant cardiac arrhythmias, pericardial effusion, or congestive heart failure, requiring hospitalization, specific procedure, or drug treatment. Scleroderma renal crisis (SRC) was defined as new-onset systemic hypertension > 150/85 mm Hg and a documented decrease in estimated glomerular filtration rate > 30% with microangiopathic hemolytic anemia on blood smear, and/or retinopathy typical of acute hypertensive crisis, and/or new onset of urinary red blood cells (excluding other causes), and/or flash pulmonary edema, and/or oliguria or anuria, and/or definitive results of a renal biopsy showing characteristic changes. Followup was calculated as the time between first non-Raynaud’s symptom of SSc and the date the patient was last reviewed, or died. The study was approved by the Royal Free Hospital local ethics committee.
Statistical analysis
Fisher’s exact test was used to compare gender, disease subset, autoantibody, and overlap syndrome frequencies between the juvenile- and adult-onset patients. Log-rank test was used to compare Kaplan-Meier estimates of survival and time to significant internal organ complications between the subjects with juvenile- and adult-onset SSc as well as the patients in the diffuse and limited subsets of the jSSc cohort. All calculations were done using the Minitab 14 statistical package.
RESULTS
From a cohort of over 2200 patients we identified 58 with childhood-onset SSc, 52 of whom were followed into adulthood. The remaining 6 patients were still children at the time of their last visit. A group of 954 patients with adult-onset SSc was used to compare disease course, organ complications, and outcome. Table 1 shows basic demographic and clinical characteristics of the 2 cohorts.
Demographic and clinical characteristics of patients with juvenile-onset and adult-onset SSc. Data are number (%).
The juvenile-onset SSc cohort
The 52 patients with jSSc who were adults at the time of data collection were included in this analysis. Of those, 17 (33%) were referred and seen in our center within 3 years of disease onset. The mean ± SD age at disease onset was 14 ± 2 years and the earliest onset of SSc we observed was in a 5-year-old. Thirty-nine of the 52 patients (75%) were women and 24 (46%) had the diffuse cutaneous subset of SSc. Nineteen (37%) patients showed overlap features of other connective tissue diseases, with polymyositis/dermatomyositis being the most common (19%, n = 10), followed by rheumatoid arthritis in 6 (12%), systemic lupus erythematosus (SLE) in 3 (6%), and Sjögren’s syndrome in 1 (2%). Autoantibody profile of the subjects demonstrated that anti-topo I antibodies were the most frequent (33%, n = 17), followed by anti-nRNP antibodies in 8 (15%). ACA and ARA were found only in 1 patient each. Similarly, only 1 patient was anti-PM-Scl antibody-positive, 1 was anti-Jo1 antibody-positive, and 2 were anti-U3RNP-positive. Nearly a quarter of the patients (23%, n = 12) had nonspecified antinuclear antibodies (ANA). Interestingly, 4 (8%) of the subjects were ANA-negative.
All subjects had skin involvement and Raynaud’s phenomenon. Analysis of internal organ disease demonstrated that, overall, PF was the most common complication, found in 28 (54%) of the patients. In 16 of them (31% of the whole cohort) PF was clinically significant, as defined above. Seven of the subjects (13%) developed PAH, 1 SRC, and 1 cardiac SSc.
Data on drug treatment were available for 44 of the 52 patients (85%) in this study. Of those, 26 (50% of the jSSc cohort) had received disease-modifying treatment at some point during their followup. The most frequently used immunosuppressive agents were methotrexate (19% of subjects, n = 10), azathioprine (17%, n = 9), mycophenolate mofetil (12%, n = 6), and cyclophosphamide (10%, n = 5). Other treatments included antithymocyte globulin, cyclosporine, stem cell transplantation, hydroxychloroquine, D-penicillamine, salazopyrin, adalimumab, and intravenous immunoglobulin. In addition, 19 (37%) had received oral and 4 (8%) intravenous corticosteroids.
Only 11 (21%) patients with jSSc were referred to us before they had reached adulthood. The mean ± SD age at first visit was 25 ± 10 years (range 13–49 yrs). The average ± SD followup of the patients with jSSc in our department was 9 ± 7 years (range 0–25). At the time of death/last visit, mean age was 35 ± 12 years (range 17–63), while mean disease duration was 21 ± 12 years (range 3 months to 51 years). At the time of data collection 11 patients (21%) were lost to followup (had not been seen for more than 2 years). Kaplan-Meier survival at 5, 10, 15, and 20 years was 100%, 98%, 95%, and 82%. Out of the 52 patients, 14 (27%) had died and causes of death were known for 10 of them. Most frequently, death was lung-related, with 3 patients dying from PAH, 2 from severe PF, 2 from lung cancer, and 1 from acute lupus pneumonitis on the background of SSc/SLE overlap, PF, and PAH. One patient died of amyloidosis and 1 of sepsis on a background of chronic osteomyelitis and chronic renal failure. There was no difference in terms of sex, disease subset, autoantibody profile, frequency of overlap syndromes, and internal organ complications between the deceased patients and the ones who were still alive at the time of data collection.
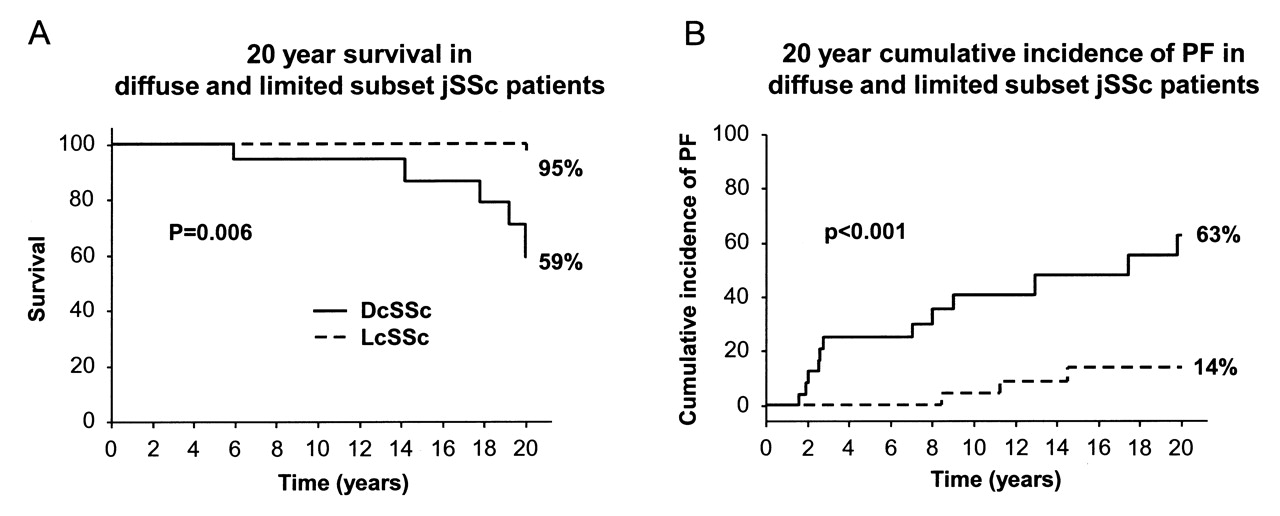
Comparison between the diffuse and limited subset patients from the jSSc cohort at 20 years from disease onset demonstrated significantly better survival (95%) and lower cumulative incidence of PF (14%) among the patients with lcSSc compared to the dcSSc group, where survival was 59% (p = 0.006), and frequency of PF was 63% (p < 0.001; Figure 1).

Comparison of survival and cumulative incidence of pulmonary fibrosis (PF) at 20 years from disease onset in diffuse and limited cutaneous subset patients from the juvenile-onset SSc cohort. DcSSc: diffuse cutaneous SSc; LcSSc: limited cutaneous SSc.
Adult and juvenile SSc cohort comparison
Comparison between the adult- and juvenile-onset SSc cohorts revealed no significant differences between the 2 cohorts in sex distribution and disease subset. There was a significantly higher proportion of patients with overlap syndromes in the jSSc cohort (37%) compared to the adult-onset group (18%; p = 0.002) and this was mainly due to the higher proportion of patients with polymyositis/dermatomyositis and rheumatoid arthritis (Table 1). The autoantibody profile also showed some differences, with the jSSc cohort having a significantly higher proportion of patients positive for anti-topo I antibodies (33%) compared to the adult-onset cohort (20%; p = 0.033), while the ACA was significantly less frequent (only 2% of the jSSc cohort) compared to 25% of the adult-onset cohort (p < 0.001).
We compared survival and clinically significant internal organ complication frequencies at 10 years from disease onset in the 2 cohorts. The Kaplan-Meier survival at 10 years among the patients with jSSc was significantly higher (98%) compared to the adult-onset group (75%; p = 0.001; Figure 2A). There was also a significantly lower incidence of PAH in the jSSc group (2%) compared to 14% in the adult-onset cohort (p = 0.032). On the other hand there was no difference between the 2 cohorts in cumulative incidence of PF (22% for patients with adult-onset and 21% for childhood-onset SSc; Figure 2B) as well as cardiac scleroderma (3% and 2%, respectively). None of the patients with jSSc developed renal crisis within the first 10 years of their disease, while this complication occurred in 7% of the adult-onset cohort (p = 0.053).

Comparison of survival and cumulative incidence of pulmonary fibrosis (PF) at 10 years from disease onset in patients with juvenile- and adult-onset systemic sclerosis (SSc).
DISCUSSION
Elucidating the differences between adult SSc cases that had childhood onset and the majority of cases in which disease develops in adulthood is of clinical importance. It is essential for both risk stratification and outcome prediction in longterm followup in the subgroup of childhood-onset cases. In our study we compared 2 cohorts of adult patients with SSc who had disease onset either during childhood or in adulthood. As expected, the jSSc group had a significantly higher proportion of patients with overlap syndromes and had a much better overall survival. While cumulative incidences of PF and cardiac disease were comparable between the 2 groups, PAH and renal crisis were much more frequent among the adult-onset SSc subjects.
The 2 largest published pediatric SSc cohorts to date were a result of international multicenter data collection in pediatric rheumatology centers in Europe, the Americas, Asia, Australia, and Africa4,8. Disease subset and autoantibody data were collected only for the PRES international database cohort4. Out of 153 subjects, 139 (91%) had the diffuse cutaneous subset of SSc, 34% of the subjects were ATA-positive, and 7% were ACA-positive. Neither study included data on overlap syndromes, and organ complications were not strictly defined. The overall conclusions from both studies suggested less severe organ disease and significantly better outcome with higher survival rates for patients with jSSc compared to adult-onset subjects.
Two other cohorts, consisting of adult jSSc subjects, were identified from the Pittsburgh Scleroderma Databank and the EUSTAR database3,5. Unlike the results of the PRES database study, these 2 adult cohorts demonstrated no difference between adult- and juvenile-onset patients in terms of subset distribution and organ complications in adulthood, with the exception of disproportionately low incidence of renal disease among the Pittsburgh patients with jSSc. Both studies also confirmed the rarity of ACA among patients with jSSc. In addition, a recent publication by Martini, et al analyzed the factors affecting survival among the patients included in the PRES database9. All patients who died had the diffuse cutaneous subset of SSc. Over 60% of the deaths occurred within 5 years from disease diagnosis and over 90% within 10 years from disease diagnosis.
Interestingly, the overall survival in our cohort (100% at 5 years, 98% at 10 years, 95% at 15 years, and 82% at 20 years) was much better than survival in the pediatric cohort from the PRES database (88% at 5 years, 85% at 10 and 15 years, and 79% at 20 years). The better survival in the adult-aged jSSc cohorts as well as the remarkable change in the proportion of dcSSc subjects from 90% in childhood cohorts to around 40% in the adult ones could represent a survival bias, suggesting that reaching adulthood may be associated with overall better prognosis.
The study by Martini et al proposed a model for prediction of outcome in patients with jSSc including fibrosis on chest radiographs (OR 11.1; p = 0.02), raised creatinine levels (OR 22.7; p = 0.02), and pericarditis (OR 41.3; p = 0.001) as the factors with the strongest association with poor prognosis9. It is difficult to compare the results of different studies in terms of organ complications, because of the very different definitions of organ disease that have been used. In our cohort we used strict criteria to define moderate to severe organ complications. Our analysis showed no significant difference in the frequencies of PF and cardiac disease between the adult- and childhood-onset groups and only a trend toward significance when comparing renal crisis frequency. Nevertheless, the 10-year survival rate was significantly better among the subjects with jSSc compared to adult-onset ones.
As expected, the proportion of patients with overlap syndromes was significantly higher among the jSSc cohort compared to the adult-onset SSc group, and this is consistent with the findings of other studies3. Similarly, the serological characteristics of our patients concurred with previous publications, demonstrating disproportionately low rates of ACA positivity, while ATA anti-U1RNP antibodies were significantly more frequent among the subjects with jSSc.
Our study has certain limitations. All the patients included were identified from a cohort and followed in an adult connective tissue disease specialist center. As a result, only a small proportion of the subjects (21%) were referred to us in their childhood, and we included in the analysis only those patients who had reached adulthood at the time of data collection. It is likely that our cohort differs from jSSc cohorts seen in pediatric centers and represents only those who have generally milder disease and survive into adulthood. We also acknowledge the drawbacks related to the retrospective design of the study, with missing data, especially that covering the period between disease onset and referral to our center. On the other hand, the derivation of the jSSc and adult-onset SSc groups from a single-center cohort, using uniform disease characteristic definitions and similar followup patterns, allowed a reliable comparison with results applicable to other adult SSc cohorts.
The high survival rates and lower proportion of patients with the diffuse subset in our cohort may represent a survival bias. Prospective multicenter cohort studies should be considered to accurately assess to what extent the differences in disease presentation and outcome that we see in adult patients with childhood disease onset as opposed to adulthood onset are due to differences in SSc course rather than loss of more severe cases.
- Accepted for publication July 14, 2010.








{kind=link}
{kind=link}