

Abstract
Objective. Patients with systemic sclerosis (SSc) have significantly fewer and functionally impaired endothelial progenitor cells (EPC) in peripheral blood and bone marrow; further, endothelial apoptosis seems to play a primary role in the pathogenesis of vascular damage. We investigated whether the failure of bone marrow EPC is related to their apoptotic phenotype and analyzed the possible mechanisms inducing apoptosis.
Methods. The presence of apoptotic cells was investigated in bone marrow aspirates taken from patients with SSc; microvessel density (MVD) and the immunohistochemical expression of vascular endothelial growth factor (VEGF) were also measured in bone marrow biopsies. A correlation between EPC apoptosis and the presence of antiendothelial cell antibodies (AECA) was also investigated.
Results. We confirmed the presence of bone marrow EPC dysfunction in SSc, while hematopoiesis was not impaired. Bone marrow studies showed a high percentage of apoptotic progenitors, no signs of fibrosis or an altered MVD, and an increased VEGF index. The patients’ bone marrow plasma showed significant titers of AECA, and their presence correlated with that of apoptotic progenitors. These findings were further confirmed by an in vitro assay in which the apoptosis of normal progenitors was induced by the addition of AECA+ purified IgG.
Conclusion. Our results showed that apoptosis in patients with SSc involves the source compartment of endothelial progenitors and correlates with AECA activity. These findings support the hypothesis that AECA may play a pathogenetic role by affecting the bone marrow EPC machinery that should repair the peripheral vascular lesions.
Systemic sclerosis (SSc) is a complex autoimmune disorder characterized by extensive fibrotic changes in various organs, including skin and lung. Although its etiology is unknown, the main pathophysiological features of SSc are the complex interactions between immune activation, vascular injury, and fibroblast dysfunction1,2.
Functional and structural vasculopathy is believed to be the primary and pivotal inducer of scleroderma tissue damage. Pathological changes in the vascular tree range from endothelial activation with an increased expression of adhesion molecules to endothelial apoptosis with intimal proliferation of arterioles and capillary necrosis and finally, blood vessel occlusion; the progressive decrease in the size of microvascular beds and reduced organ blood flow ultimately lead to chronic tissue ischemia3. However, although the pathological course of SSc vasculopathy is well known, the mechanisms that start the vascular damage and/or prevent its repair remain to be established. A number of studies have pointed out that the vascular endothelium may be the primary target of the autoimmune aggression4,5; more specifically, it is thought that antiendothelial cell antibodies (AECA) mediate endothelial cell injury and induce apoptosis6, as has been shown in the UCD-200 chicken model of SSc and further confirmed by the in vitro demonstration of endothelial cytotoxicity7,8,9,10,11.
In addition to the established importance of immune-mediated endothelial aggression in the development of scleroderma vascular disorder, there is growing evidence that an impaired vascular repair mechanism may also play an important role in the vascular “drop out.” Many studies during the last 10 years have found that circulating bone marrow-derived cells can form new blood vessels by means of a process of postnatal vasculogenesis, with endothelial progenitor cells (EPC) being selectively recruited to injured or ischemic tissue12. A number of authors have shown that patients with SSc seem to have defects in the number and function of circulating EPC, thus providing additional support for the hypothesis that EPC play a pathogenetic role in the defective vasculogenesis associated with the disease13,14,15,16,17. Because it has been demonstrated that the numerical and functional impairment of EPC lies in the bone marrow of patients with SSc14,17, it has been hypothesized that SSc vasculopathy may be the result of an inability to repair and form new blood vessels.
Our aim was to examine the possibility that the immuno-mediated mechanisms capable of inducing peripheral endothelial injury in SSc (i.e., apoptotic phenomena and/or antiendothelial activity) are also found in the bone marrow environment.
MATERIALS AND METHODS
Patients
The study involved 28 patients fulfilling the SSc criteria of the American College of Rheumatology18, who were classified as having limited (lcSSc) or diffuse (dcSSc) SSc on the basis of LeRoy’s criteria19. Any patients whose symptoms overlapped those of other connective diseases were excluded, as were pregnant women, smokers, people with diabetes, hypertensive subjects, patients with significant hyperlipidemia, and those receiving cholesterol-lowering medications, including statins.
The study population was clinically assessed on the basis of described criteria20,21. For all patients with SSc, disease activity was assessed using the activity indexes of the European Scleroderma Study Group22,23. Sixteen of the 28 patients were being treated with intravenous prostanoid. Bone marrow samples were obtained just before the iloprost infusion and at least 1 month after the previous one. Angiotensin-converting enzyme inhibitors and calcium channel blockers were discontinued at least 3 weeks before the study, and none of the patients had received any disease-modifying drugs (i.e., high dosages of corticosteroids, cyclophosphamide, or methotrexate).
The control group consisted of 20 healthy donors of bone marrow for allogeneic transplantation (median age 48 years, range 28–55).
This study was approved by the local ethics committees; all the participants gave their written informed consent.
Bone marrow biopsies
Formalin-fixed, paraffin-embedded bone marrow biopsies (BMB) obtained from the posterior superior iliac spine were available for 14 patients with SSc.
BMB were decalcified using an EDTA-based solution (33.27 g EDTA and 10 ml hydrochloride diluted in 1 liter distilled water) for 4 h, and sections from each block were routinely stained with hematoxylin-eosin, Giemsa stain, and Gomori’s silver impregnation.
Immunohistochemistry was performed using the automated Genomix i-6000 staining system (BioGenex, San Ramon, CA, USA) and CD34 (dilution 1:100, clone QB-END/10; Dako A/S, Glostrup, Denmark), vascular endothelial growth factor (VEGF; dilution 1:400, rabbit polyclonal antibody; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and CD117 antibodies (polyclonal; Dako A/S). Heat-induced antigen retrieval was performed using a 0.01 M citrate solution at pH 6.0 in a microwave oven at 750 W (2 cycles × 5 min). The reaction was revealed by means of the Dako ChemMate EnVision Detection Kit (Dako A/S) in accord with the manufacturer’s instructions. The negative control slides were incubated with normal goat serum.
Microvessel density (MVD) was evaluated by means of CD34 immunostaining using the “hot spots” method (MVD-HS) in which each BMB is first totally scanned at low magnification (10×) to identify 3 “hot spot” zones, after which MVD-HS is estimated at high magnification (40×) by counting all of the positively stained endothelial cells or endothelial cell aggregates that are clearly separate from adjacent microvessels; sinusoid-like structures (but not arterioles) were also counted24.
We also evaluated the expression of VEGF, the most important proangiogenic factor known to be involved in the differentiation, proliferation, and mobilization of EPC. To avoid any bias related to variations in the patients’ BMB cellularity, we calculated a VEGF expression index, defined as the cellularity of the BMB multiplied by the fraction of VEGF-positive cells, and expressed as a number between 0 and 1 [(percentage of BMB cellularity × percentage of VEGF-positive cells)/104], as described25.
Bone marrow morphology, MVD, and VEGF immunohistochemical expression were evaluated by 2 pathologists experienced in evaluating bone marrow specimens and who were blinded to the clinical data and diagnosis.
Isolation of bone marrow low-density mononuclear cells
An average 10–15 ml of bone marrow obtained from each patient was collected in heparinized tubes and layered on a Ficoll-Paque gradient (specific gravity 1.077 g/ml; Nycomed Pharma AS, Oslo, Norway); the low-density mononuclear cells (LDMNC) were resuspended in Iscove’s modified Dulbecco’s medium (IMDM; BioWhittaker, Caravaggio, Italy) supplemented with 10% fetal bovine serum (FBS; BioWhittaker).
Bone marrow CD133+ cell separation
The hematopoietic stem and progenitor cells were isolated using a positive selection of CD133-expressing cells. Bone marrow LDMNC were incubated 30 min with the AC133/1 monoclonal antibody (mAb) directly labeled to microbeads (MACS; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany), washed, filtered through a 50-μm nylon mesh to remove clumps, and placed on a column in the midiMACS cell separator (Miltenyi Biotec). The labeled cells were separated using a high-gradient magnetic field, and eluted from the column after their removal from the magnet. The positive fraction was then placed on a new column and the magnetic separation step repeated. At the end of the separation, the cells were counted and assessed for viability using Trypan Blue dye exclusion; their purity was determined using a FACSCanto II flow cytometer and Diva software (Becton Dickinson, Milan, Italy).
A total of 3 × 105 CD133+ enriched cells were plated on fibronectin-coated (Boehringer Mannheim, Milan, Italy) 25-T tissue culture flasks, grown in M199 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FBS, 50 ng/ml VEGF, 1 ng/ml basic-fibroblast growth factor and 2 ng/ml insulin-like growth factor-1 (Peprotech, London, England), and cultured as reported26.
Flow cytometry
A total of 2.5 × 105 bone marrow LDMNC were incubated 20 min at 4°C with selected primary mAb against CD34-FITC (8G12), CD3-phycoerythrin (PE), CD19-allophycocyanin (APC; SJ25C1), CD45-PerCP (2D1; all from Becton Dickinson), AC133/2-PE and AC133-APC (293C3; CD133; Miltenyi Biotec, Calderara di Reno, Italy), and kinase domain receptor (KDR)-APC (89106; R&D Systems, Minneapolis, MN, USA), and then washed and evaluated using a FACSCanto II flow cytometer and Diva software.
For IgG binding evaluation, a total of 2.5 × 105 bone marrow LDMNC were incubated 20 min at 4°C with the primary antibodies against AC133/2-PE, CD45-PerCP, and human IgG-APC. The cells were then washed and evaluated using the FACSCanto II flow cytometer and Diva software.
Progenitor assays
Lineage-committed hematopoietic progenitors in the bone marrow were quantified on the basis of their growth in semisolid cultures as colony-forming units for granulocytes and macrophages (CFU-GM) and burst-forming units for erythroid cells (BFU-E). The assays were carried out by plating 5 × 104 bone marrow LDMNC in a methylcellulose culture medium (MethoCult GF H4434; StemCell Technologies Inc., Vancouver, BC, Canada) containing 0.9% methylcellulose in IMDM, 30% FBS, 1% bovine serum albumin, 10−4 M 2-mercaptoethanol, 2 mM L-glutamine, 50 ng/ml recombinant human stem cell factor, 10 ng/ml recombinant human granulocyte macrophage-colony stimulating factor, and 10 ng/ml recombinant human interleukin 3, 3 U/ml recombinant human erythropoietin, and incubating triplicate dishes at 37°C and 5% CO2 in a fully humidified atmosphere. After 14 days of culture, aggregates of ≥ 40 cells were scored as colonies and counted27,28.
Apoptosis in bone marrow cells
To investigate the presence of apoptotic cells among the bone marrow endothelial progenitors, we used the annexin-V-fluorescein isothiocyanate conjugated/propidium iodide (PI) assay (Apoptosis Detection Kit I; Becton Dickinson), according to the manufacturer’s instructions. Annexin-V binds with high affinity the membrane phosphatidylserine translocated to the cell surface soon after the induction of apoptosis.
A total of 2.5 × 105 bone marrow LDMNC were incubated with the primary mAb CD133-APC or KDR-APC, washed twice with cold phosphate buffer solution (PBS), and then resuspended in 100 μl 1× annexin binding buffer. They were then incubated with 5 μl Annexin V-FITC and 5 μl PI for 15 min at room temperature in the dark, and washed with 1× annexin binding buffer. A total of 100,000 events were acquired per sample, and analyzed within 1 h using the FACSCanto II flow cytometer and Diva software.
Apoptotic CD133+ or KDR+ cells were sequentially gated as low side- or forward-scattered in the stem cell area, excluding cell debris, and apoptosis was estimated as the sum of the percentage of cells in early apoptosis (annexin-V+/PI-negative) plus the percentage in late apoptosis (annexin-V+/PI+)29.
Endothelial cell cultures and AECA assay
Human microvascular dermal endothelial cells (HMVEC-d; Clonetics Corp., San Diego, CA, USA) were cultured with microvascular endothelial cell growth medium (EGM-2-MV Bulletkit; Clonetics Corp.). AECA were detected using a cell-surface ELISA on confluent living cells, as described30. The optical density (OD) value of a positive reference serum at standard 1:25 dilution was arbitrarily defined as 100% of endothelial binding activity. The results of tested samples were expressed as a percentage of this positive reference value, to adjust for interassay variability. An OD > 3 SD above the OD obtained with sera from 50 healthy individuals (i.e., 35% for AECA IgG and 48% for AECA IgM) was considered positive31,32.
Immunofluorescence detection of AECA on CD133+ cells
Bone marrow CD133+ cells were isolated from 3 patients with SSc positive for bone marrow plasma AECA by immunomagnetic selection (Miltenyi Biotec, Bergisch Gladbach, Germany), as described14. The isolated CD133+ cells were cytocentrifuged onto glass slides (Shandon Cytospins II Cytocentrifuge, Thermo Fisher Scientific Inc., Waltham, MA, USA) and analyzed by means of immunofluorescence studies for the presence of surface IgG.
The cells were incubated for 20 min with PBS containing 20% human decomplemented AB serum and 1% bovine albumin, labeled with a mouse antihuman CD133 mAb (Miltenyi Biotec) for 30 min at room temperature, washed with PBS, and then incubated with conjugated polyclonal rabbit antimouse immunoglobulins/TRITC conjugated (Dako Italia, Milan, Italy) for 30 min at room temperature. After washing, the cells were directly labeled with polyclonal antihuman IgG/FITC conjugated (Dako Italia) for 30 min at RT, and then washed and counterstained with 4’,6-diamidino-2-phenylindole for 5 min at RT. Fluorescent images were taken with a Nikon Eclipse 50i fluorescent microscope.
Purification of polyclonal IgG
The IgG fractions from SSc AECA+, SSc AECA-negative, and normal control plasma were affinity-purified by HiTrap Protein G column (GE Healthcare, Uppsala, Sweden) in accord with the manufacturer’s instructions, and the final IgG concentration was evaluated by nephelometry.
In vitro induction of endothelial cell apoptosis
The effect of AECA-IgG on bone marrow progenitors was tested using fresh LDMNC from normal bone marrow, which were incubated with polyclonal IgG purified from 2 AECA+ patients, 2 AECA-negative patients, and 1 healthy control (at a final concentration of 5 × 104 cells/50 μg of purified IgG), and incubated for 24 h in IMDM and 5% FBS at 37°C and 5% CO2. Apoptosis was detected using the Annexin V assay and flow cytometry analysis for the expression of both CD133 and KDR, as described. Baseline cell apoptosis was quantified after 24 h without IgG treatment.
Statistical analysis
The bone marrow cell-surface marker levels, clonogenic activity, and effects of AECA on bone marrow progenitors were compared using Student’s 2-tail t-test for unpaired data.
The correlations between the number of apoptotic progenitors and AECA levels were assessed using Spearman’s rank correlation test. A p value < 0.05 was considered significant.
RESULTS
Patient demographic and clinical characteristics
The study involved 26 women and 2 men (median age 55 yrs; range 21–60): 11 were classified as having lcSSc and 17 as having dcSSc; the median disease duration was 5.0 years (range 1–29). On the basis of Medsger and Steen’s criteria21, 5/11 patients with lcSSc (45.4%) and 10/17 with dcSSc (58.8%) had early disease. Raynaud’s phenomenon was recorded in all patients. Cutaneous ulcers were found in 10 patients (35.7%). Seven patients had primary pulmonary hypertension and 8 had pulmonary fibrosis with or without pulmonary hypertension.
The evaluation of disease activity according to the European Scleroderma Study Group criteria indicated that 10 out of 28 patients had active disease.
Bone marrow biopsy evaluation
In order to investigate whether an altered vascularization and/or the presence of fibrosis could be responsible for the impairment of bone marrow progenitors, we analyzed bone marrow biopsies from patients with SSc.
Bone marrow cellularity was within normal limits33, and the 3 hematopoietic lineages were quantitatively well represented. A slight degree of dyserythropoiesis (increased precursors with left shifting and abnormalities in the topographical distribution of erythrocytes) and dysmegakaryopoiesis (increased megakaryocytes with hypolobulated and hyperchromatic nuclei, and the presence of scattered micromegakaryocytes) was found in 92% of the BMB.
There was no evidence of bone marrow fibrosis.
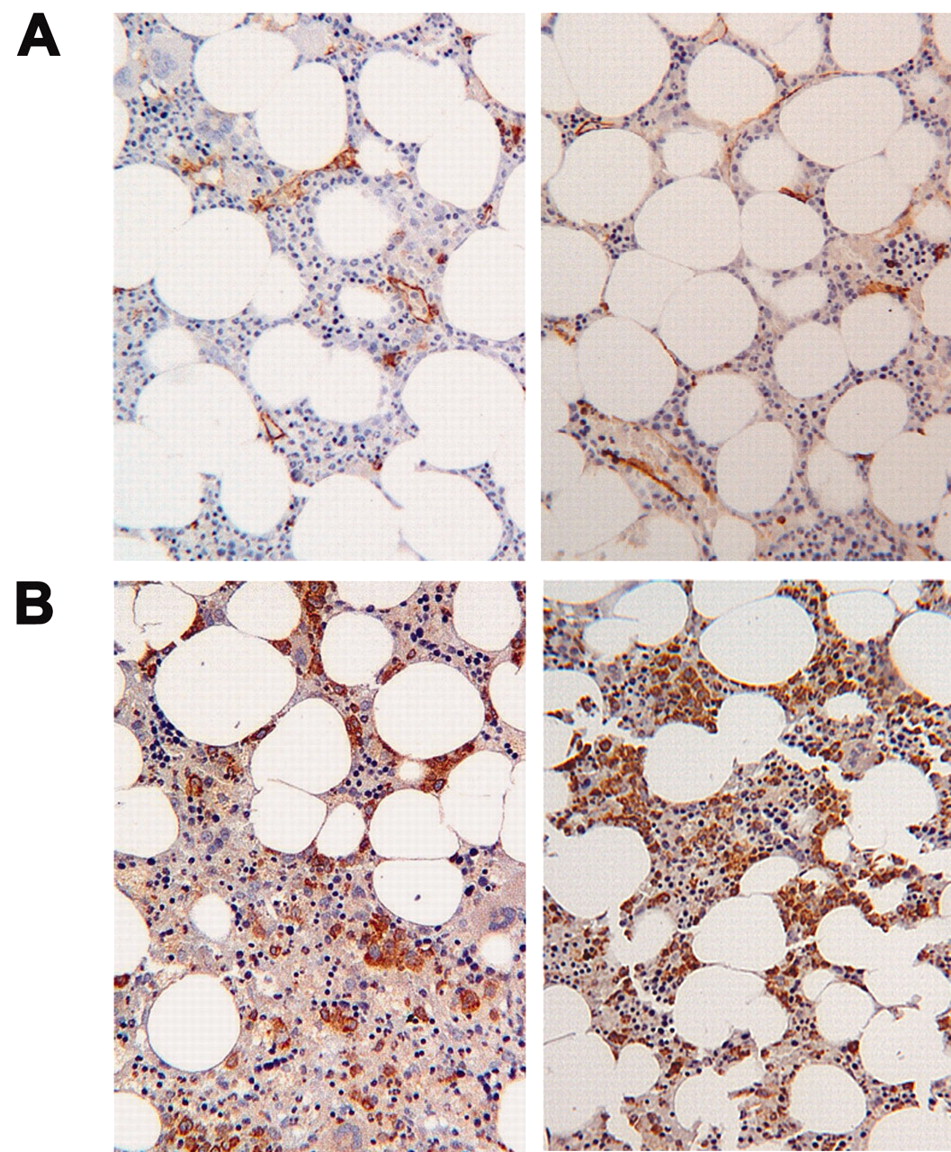
There were no statistically significant differences in MVD between the patients with SSc and the controls (MVD-HS 28.3 ± 5.6 vs 24.4 ± 2.3; Figure 1A).

(A) Systemic sclerosis (SSc) bone marrow biopsy (BMB) morphological evaluation. Microvessel density was evaluated by CD34 immunostaining using the “hot spots” method in healthy (left) and SSc (right) BMB. (B) Vascular endothelial growth factor (VEGF) index evaluation in SSc BMB. The index was defined as the cellularity of the BMB multiplied by the fraction of VEGF-positive cells, and was evaluated in BMB from patients with SSc (right) and healthy controls (left).
The expression of VEGF, the most important proangiogenic factor involved in the differentiation, proliferation, and mobilization of EPC, was significantly higher in the bone marrow of the patients with SSc (VEGF index: 0.18 ± 0.04 vs 0.13 ± 0.04; p = 0.04; Figure 1B), as observed in peripheral blood.
Phenotype analysis of bone marrow LDMNC
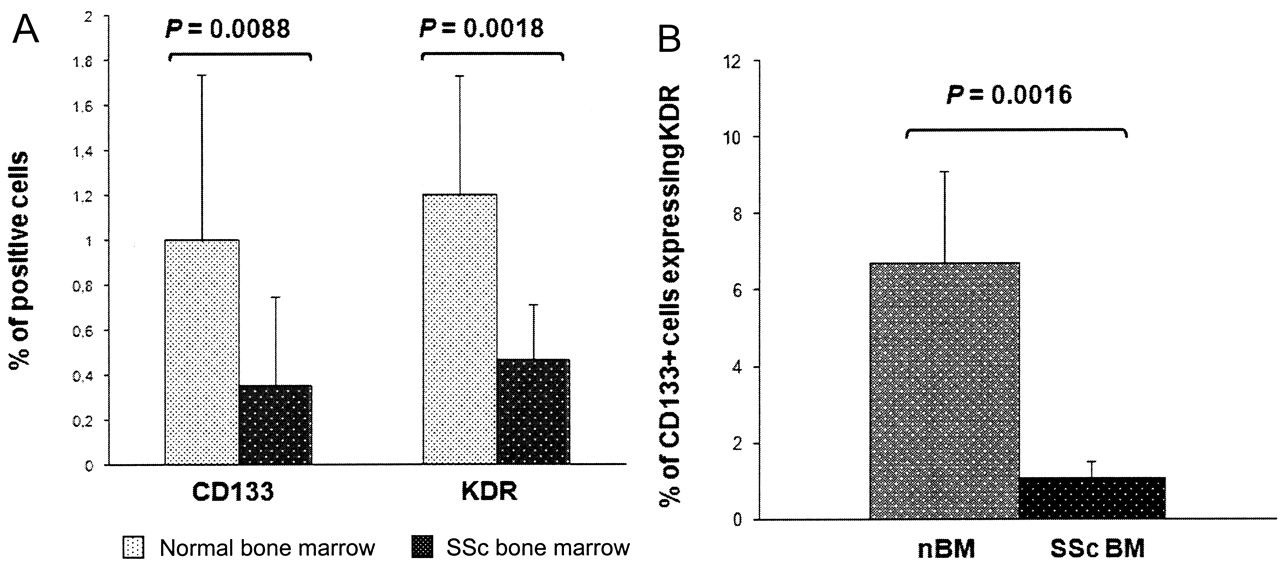
With regard to endothelial stem cell sources, the SSc bone marrow showed significantly lower percentages of CD133+ (0.35 ± 0.4% vs 1 ± 0.74% in controls; p = 0.0088) and KDR+ cells (0.47 ± 0.24% vs 1.2 ± 0.53%; p = 0.0018; Figure 2A), and showed significantly less CD133/KDR coexpression: 0.002 ± 0.0003% vs 0.061 ± 0.022% (p = 0.013). By evaluating the whole CD133+ cell population, only 1.1 ± 0.41% cells expressed KDR positivity in SSc (vs 6.71 ± 2.4% in normal bone marrow; p = 0.016; Figure 2B).
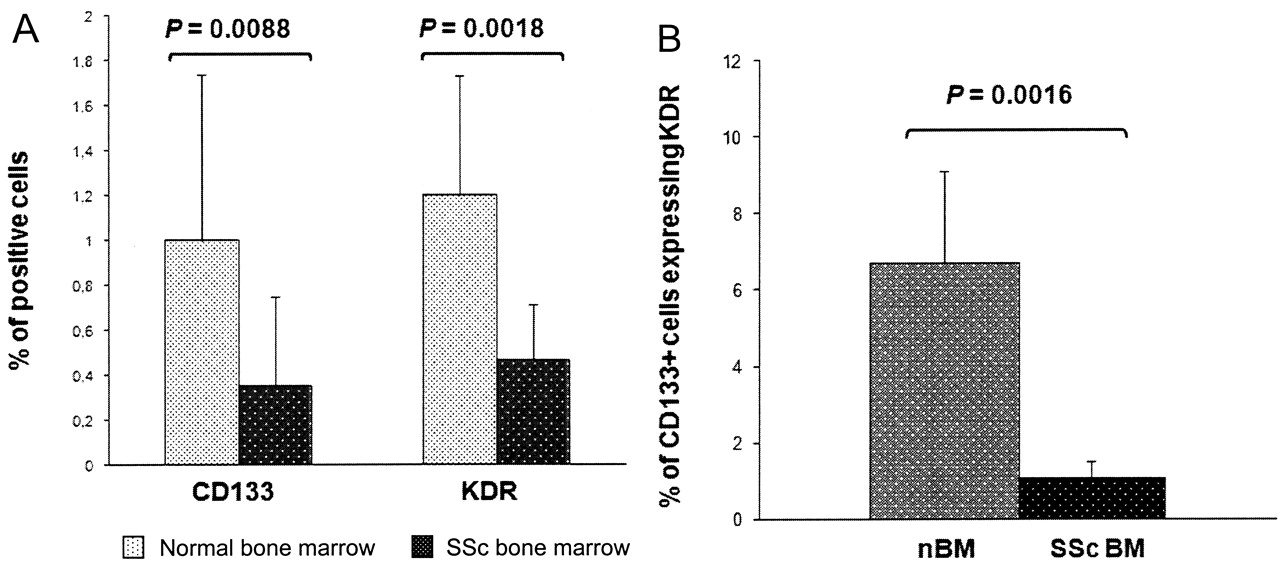
Flow cytometry evaluation of bone marrow (BM) endothelial progenitors. (A) Comparison of bone marrow CD133 and KDR expression in patients with SSc and controls evaluated by flow cytometry. p < 0.05 was considered significant. (B) Percentage of CD133+ cells coexpressing KDR in patients with SSc and controls (normal bone marrow, nBM).
Hematopoietic progenitors
In order to investigate if SSc bone marrow CD133+ cell impairment is generalized to the whole stem cell compartment or selective for the endothelial-lineage progenitors, we quantified lineage-committed hematopoietic progenitors according to their growth in semisolid cultures: CFU-GM and BFU-E27,28. There was no significant difference in the numbers of hematopoietic progenitors between the patients and controls (CFU-GM: 92 ± 46.6 vs 123 ± 45; BFU-E: 124.7 ± 68.5 vs 136.4 ± 47).
CD133+ cell separation and endothelial differentiation
The mean recovery of CD133+ cells after immunomagnetic separation was 0.11 ± 0.04%, lower than in the controls (0.35 ± 0.18%), with a purity of 82 ± 6.8% as determined by flow cytometry, confirming our data14.
In order to induce the endothelial differentiation of separated bone marrow progenitor cells, we cultured the CD133+ cells as reported26. The cells were grown on fibronectin-coated flasks in the presence of VEGF, basic fibroblast growth factor, and insulin-like growth factor-1. After 21 days of culture, only rare cells had formed small colonies that did not expand, and the cells rapidly showed aging and suffering and died.
Apoptosis of CD133+ and KDR+ bone marrow progenitors
The mean percentage of apoptotic CD133+ cells was 31.8 ± 13% in the patients and 13.08 ± 7.86% in the controls (p = 0.016); the corresponding figures for KDR+ cells were 45.8 ± 16.7% and 24.3 ± 14.9% (p = 0.02; Figure 3). The difference in the percentages of apoptotic CD133+ and KDR+ cells in the patients with SSc was also statistically significant (p = 0.017). By contrast, low overall rates of apoptosis were found in the LDMNC fraction as a whole in both patients and controls (13.3 ± 7.4% vs 10.7 ± 1%). No correlations were found between apoptotic phenomena and the subsets of the disease, activity score, or organ involvement.

Bone marrow progenitor apoptosis. Apoptotic CD133+ or KDR+ cells were sequentially gated as low side- or forward-scattered in the stem cell area, and apoptosis was estimated as the sum of the percentage of cells in early apoptosis (Annexin-V+/PI-negative) plus the percentage in late apoptosis (annexin-V+/PI+). Legends show the percentages of apoptotic cells on the whole population (TOT APOP), of CD133+ or KDR+ cells, and of CD133+ or KDR+ apoptotic cells. (A) Representative panel showing CD133+ cell evaluation in healthy (left) and SSc (right) bone marrow. Total bone marrow cell population in red. Apoptotic cells in green. CD133+ cells in blue. Apoptotic CD133+ cells in purple. Panels indicate the percentages of total apoptosis, CD133+ cells, and apoptotic CD133+ cells in reference to the whole population of bone marrow low-density mononuclear cells (% Total Pop) and the percentages of apoptotic CD133+ cells in reference to the whole CD133+ population (% CD133+). (B) Representative panel showing KDR+ cell evaluation in healthy (left) and SSc (right) bone marrow. Total bone marrow cell population in red. Apoptotic cells in green. KDR+ cells in blue. Apoptotic KDR+ cells in purple. Panels indicate the percentages of total apoptosis, KDR+ cells, and apoptotic KDR+ cells in reference to the whole population of bone marrow low-density mononuclear cells (% Total Pop) and the percentages of apoptotic KDR+ cells in reference to the whole KDR+ population (% KDR+). (C) Mean percentage ± SD of apoptotic CD133+ and KDR+ cells in bone marrow samples from patients with SSc and healthy controls (normal bone marrow, nBM). Data compared using Student’s 2-tail t-test for unpaired data; p < 0.05 considered significant.
Prevalence and clinical association of AECA in patients with SSc
SSc and normal sera and bone marrow plasma samples were evaluated for the presence of AECA. As shown in Figure 4A, AECA levels were significantly higher in the patient group in comparison to controls (% binding: 42 ± 17% vs 16 ± 5%; p = 0.0017). We found the same prevalence of AECA binding in bone marrow and peripheral plasma (data not shown). Among the 28 patients with SSc, AECA IgG were detected in 11 (39.3%) and AECA IgM in 8 (28.6%). No significant difference was observed in the prevalence of AECA positivity between patients with dcSSc and lcSSc. Further, no statistically significant correlations were found between the presence of AECA and pulmonary function results, lung fibrosis evaluated by HRTC, pulmonary artery pressures, and skin score. In contrast, a significant positive correlation was found between AECA and digital ulcers (p = 0.003).
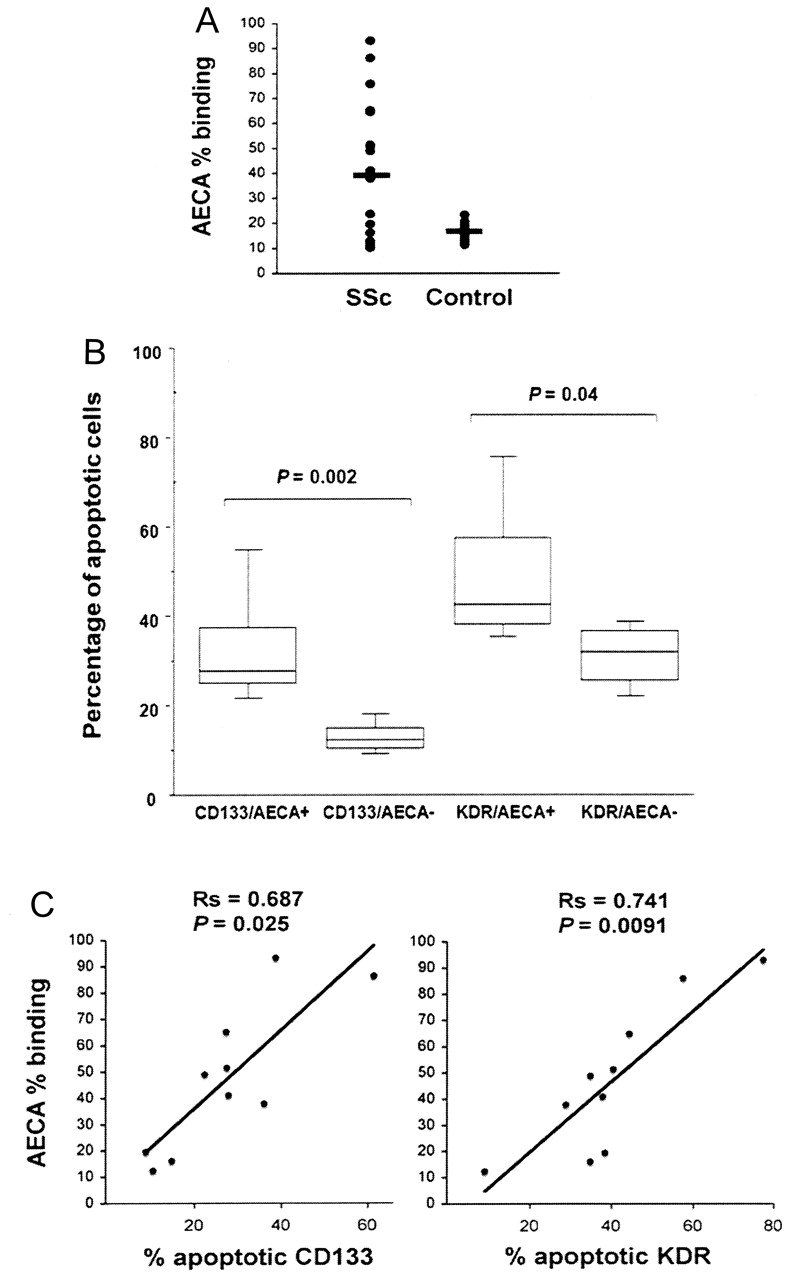
(A) Antiendothelial cell antibody (AECA) levels in bone marrow plasma of patients with SSc and controls. AECA were detected using a cell-surface ELISA on confluent living human microvascular dermal endothelial cells. Values are expressed as the percentage of binding activity. Bars correspond to median values (p = 0.0017). (B) Distribution of bone marrow CD133+ and KDR+ apoptotic cells was analyzed in AECA+ and AECA-negative subgroups of patients. In each box, lower and upper bars indicate the minimum and maximum data values; the box contains the middle 50% of the data, the upper edge indicating the 75th percentile, the lower edge the 25th percentile; bar indicates median value. (C) Correlation between bone marrow apoptotic progenitors and AECA titers. Presence of a positive correlation between AECA titers (expressed as percentage of binding activity) and number of apoptotic CD133+ or KDR+ cells was investigated using Spearman’s rank correlation test. p < 0.05 considered significant.
The percentages of apoptotic CD133+ and KDR+ cells were significantly higher in the AECA+ group of patients (32.9 ± 13% vs 12.9 ± 3.5% in the AECA-negative group, p = 0.002; 48.8 ± 16% vs 31 ± 7.3%, p = 0.04, respectively; Figure 4B); there were no significant differences between apoptotic values in the AECA-negative groups and in controls. In addition, there was a close correlation between AECA titers and the number of apoptotic progenitor cells, which was closer in relation to KDR+ cells (Rs = 0.741, p = 0.0091) than to CD133+ cells (Rs = 0.687, p = 0.025; Figure 4C).
IgG detection in SSc bone marrow cells
To highlight the binding of IgG to bone marrow progenitor cells, the CD133+ cells from the SSc and normal bone marrow samples were immunoseparated and analyzed by means of immunofluorescence studies for the presence of surface IgG. As shown in Figure 5, only the CD133+ cells from AECA+ patients stained positively (panel A); CD133+ cells from healthy controls did not show any positivity for IgG (panel B).

Surface IgG binding on SSc CD133+ bone marrow cells. CD133+ cells from SSc (A, magnification ×400) and healthy (B, magnification ×200) bone marrow samples were immunoseparated and analyzed by immunofluorescence studies for the presence of surface IgG. CD133+ cells are red; nuclei counterstained with 4’,6-diamidino-2-phenylindole are blue; IgG binding is green. (C) IgG expression on the whole low-density mononuclear cell population and IgG bound to CD133+ cells were evaluated by FACS analysis.
SSc and normal bone marrow samples were analyzed for the presence of IgG: IgG binding on the whole SSc LDMNC population was evaluated as 1.97 ± 1.4%, while IgG bound to CD133+ cells was 5.76 ± 4.5%. IgG binding was not detectable on normal bone marrow LDMNC (Figure 5C).
Apoptosis-inducing effect of AECA on progenitor cells
IgG fractions were affinity purified from 2 AECA-positive and 2 AECA-negative patients with SSc, and from 1 healthy control. All IgG preparations from AECA+ sera retained their antiendothelial binding activity until a protein concentration of 5 μg/ml was reached (data not shown). In contrast, IgG fractions from the AECA-negative patients with SSc and healthy controls did not display any significant endothelial binding in the assay.
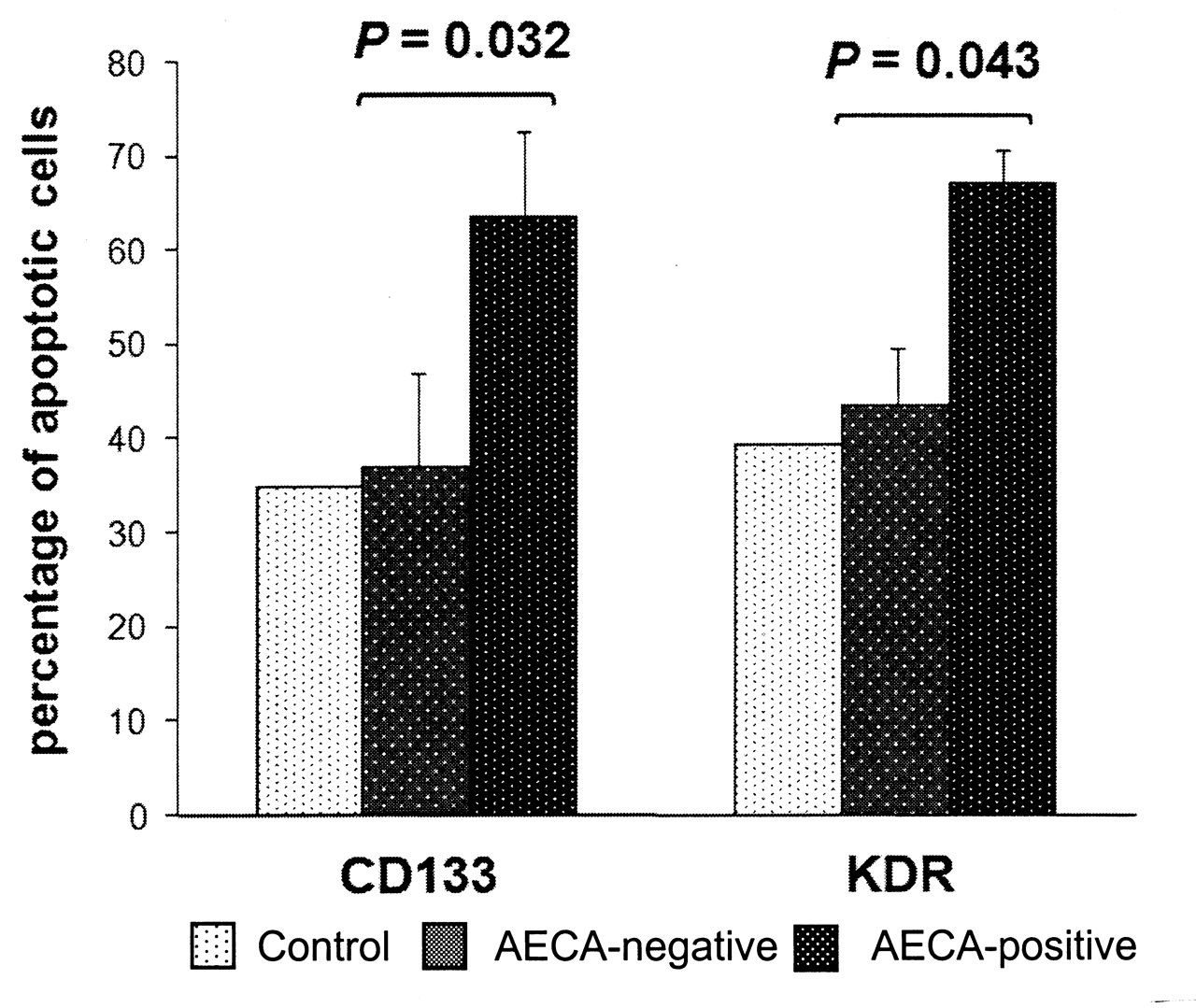
The ability of AECA to induce apoptosis in bone marrow progenitor cells was studied in an in vitro assay in which healthy bone marrow LDMNC were incubated with IgG fractions purified from AECA+, AECA-negative, and healthy control plasma samples. The incubation of AECA+ IgG fractions with normal bone marrow cells led to significantly higher mean apoptosis values than for healthy control and AECA-negative IgG fractions (Annexin V+ CD133+ cells: 35% in controls vs 37 ± 9.9% in the AECA-negative group vs 63.5 ± 9.2% in the AECA+ group, p = 0.032; Annexin V+ KDR+ cells: 39.4% vs 43.5 ± 6% vs 66.9 ± 3.7%; p = 0.043; Figure 6).
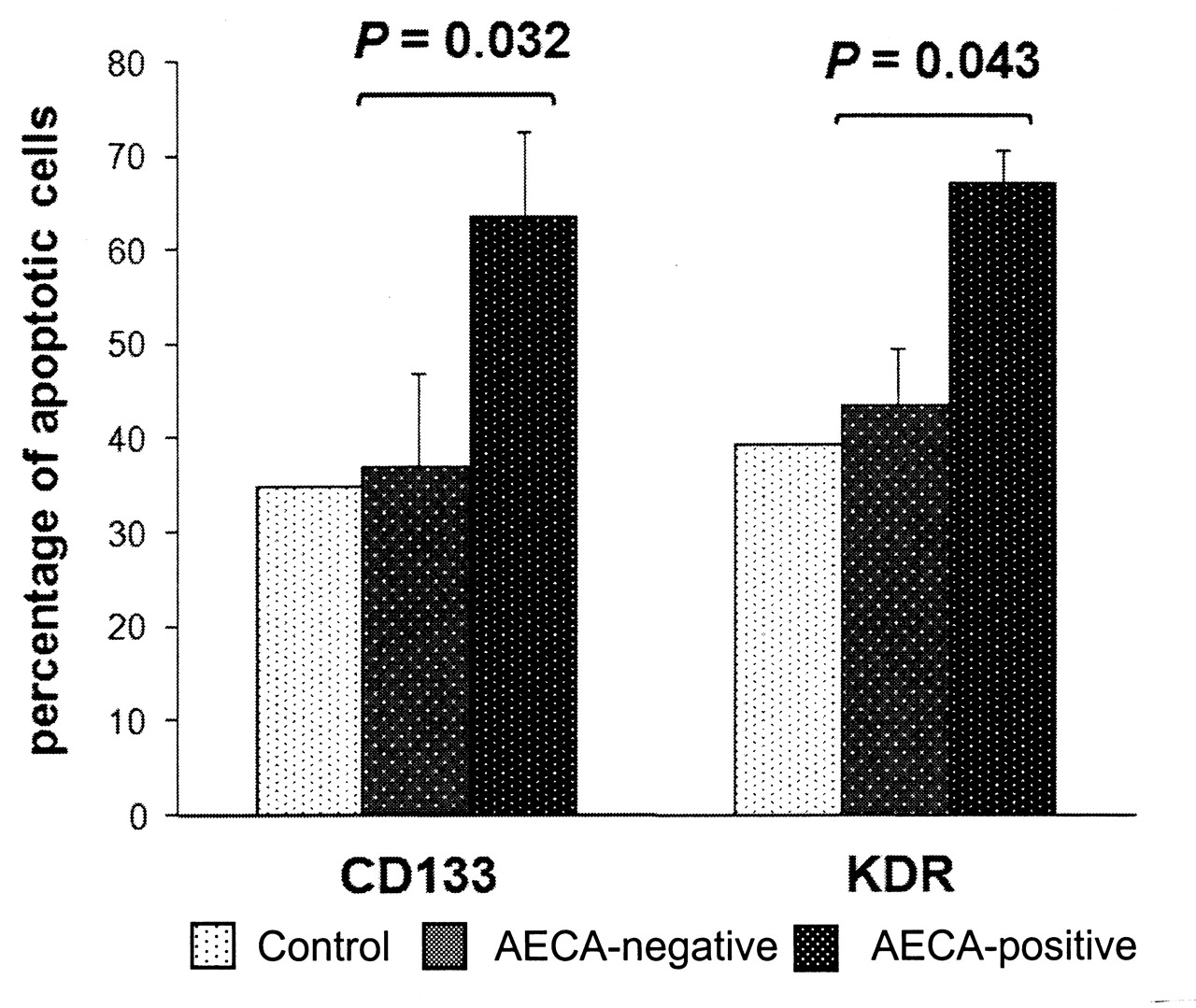
In vitro induction of apoptosis by antiendothelial cell antibodies (AECA). Fresh low-density mononuclear cells from normal bone marrow were incubated with polyclonal IgG purified from 2 AECA+ patients, 2 AECA-negative patients, and 1 healthy control (at final concentration 5 × 104 cells/50 μg purified IgG). Apoptosis was detected using the Annexin V assay and flow cytometry analysis for expression of both CD133 and KDR. Baseline cell apoptosis was quantified after 24 hours without IgG treatment and subtracted from the final values.
DISCUSSION
Recent studies have highlighted that adequate endothelial regeneration is crucial to ensuring sufficient neovascularization and tissue remodeling under conditions of chronic ischemia12,34. Thus, in the case of SSc, an important pathogenic moment may be the failed recruitment and incorporation of vascular progenitor cells into ischemic tissues. Studies have found fewer and functionally impaired EPC in patients with SSc13,14,15,16,17, but the real cause of this deficiency is unknown.
We analyzed the possible mechanisms responsible for defective vasculogenesis in SSc by investigating the pathological and functional endothelial features in the bone marrow. Our results confirm, in a larger series of patients with SSc, previous data showing that bone marrow is quantitatively deficient in EPC14, and that apoptotic phenomena involve endothelial progenitors just in their original compartment. Our findings suggest that (1) EPC impairment is not related to fibrosis or microvascular damage; (2) the bone marrow of patients with SSc contains antibodies with antiendothelial activity; and (3) IgG with antiendothelial activity can react with the surface and induce the apoptosis of CD133+ cells taken from healthy subjects.
Data on vasculogenesis in patients with SSc are still controversial, since some studies reported increased levels of circulating EPC while others described reduced EPC counts, depending on how and when EPC were phenotypically evaluated13,14,15,16,17. To date there are no specific or proven markers to identify endothelial progenitor cells and recent studies do not confirm that the well known combination of CD34, CD133, and KDR markers identifies the real endothelial precursors35,36. We showed that the bone marrow of patients with SSc contained many fewer and functionally impaired EPC, evaluated as CD133+ cells. There is evidence to support that these cells represent adult “hemangioblasts,” since many authors succeeded in differentiating endothelium from CD133+ cells26,37,38,39. CD133+ cells are multipotent hematopoietic stem cells that act as progenitors for both hematopoietic and endothelial cells. Our analysis of bone marrow from SSc clearly shows that CD133+ cells are defective only in endothelial differentiation. In contrast, the evaluation of the hematopoietic compartment shows that bone marrow cellularity is within normal limits, the 3 hematopoietic lineages are quantitatively well represented, and the number of lineage-committed hematopoietic progenitors is normal.
That fewer and functionally impaired EPC are found in the bone marrow of patients with SSc could imply that the altered production, mobilization, or half-life of EPC may play an important role in the pathogenesis of vasculopathy in SSc. Various mechanisms can be suggested to explain these findings. The exhaustion of a presumably finite supply of EPC due to their continuous peripheral recruitment to repair endothelial damage may contribute to their smaller number, as it has been suggested that EPC depletion is a risk factor in the pathogenesis of cardiovascular diseases40. Another hypothesis is the occurrence in bone marrow of microvascular abnormalities, autoimmune processes, and fibrogenesis and tissue atrophy, which are believed to be the hallmarks of the SSc disease process.
Our study clearly demonstrates that the bone marrow of patients with SSc shows neither abnormalities in vascularization nor evidence of fibrosis. We did find that the immunohistochemical expression of VEGF is higher in bone marrow biopsy specimens obtained from patients with SSc than in those obtained from controls, which is in line with data showing increased VEGF levels in the serum and skin of patients with SSc14,41,42,43,44,45. One explanation is that progressive VEGF upregulation through the various stages of the disease14 may represent an attempt to restore sufficient endothelium perfusion and integrity.
The recently described defective expression of EPC VEGFR-1 induced in vitro by hypoxia46 is another possible mechanism explaining the overexpression of VEGF in bone marrow.
Comparative studies of skin biopsy tissues from UCD-200 chickens and humans with SSc have identified endothelial activation and apoptosis as the primary pathogenetic moment in the development of SSc as it occurs before any other alterations6. Our study identifies for the first time the presence of a large number of apoptotic progenitors in the bone marrow of patients with SSc. An apoptotic phenotype was detectable in the CD133+ fraction, and was even more pronounced in the KDR+ fraction, which represents the stem cell population that is most committed to the endothelial lineage; further, the low rates of apoptosis detected in the LDMNC fraction as a whole confirm the selective nature of the apoptosis. Taken together, these findings indicate that apoptosis is a central event in SSc that involves both mature endothelial cells in the peripheral microcirculation and EPC in the bone marrow, thus mediating peripheral vascular damage and affecting vascular repair.
The mechanisms of endothelial apoptosis in SSc are not known; however, the results of many studies suggest that antibodies against the endothelial surface may play a role in inducing vascular apoptosis in patients with SSc and UCD-200/206 chickens, the only animal model of SSc5,6,7,8,9,10,11. More recently, Zhu, et al have shown that apoptosis is induced when CD133+ cells are incubated with serum from patients with SSc, and that it disappears when the serum is depleted of IgG, which, albeit only indirectly, suggests the involvement of autoantibodies in cell death47.
AECA have been reported in SSc and their prevalence varied according to the disease subset and the methodological approach that has been used in different studies48,49,50,51,52,53. In this respect, it is important to remember that research on AECA is still hampered by nonstandardized methods of detection54. The method used in this study is the one most widely employed. In addition, we found the same prevalence of IgG-AECA that was described in other SSc populations31,48. Further, it has been shown that AECA levels in patients with SSc correlate with disease severity and vascular involvement, digital ischemia, and pulmonary arterial hypertension31,50. Indeed, it seems that AECA are not a simple epiphenomenon of a previously induced vascular insult, as they can modulate endothelial functions by upregulating the surface expression of adhesion molecules and increasing leukocyte adhesion55, and induce apoptotic effects on the microvasculature5,8. We confirmed a strong correlation between AECA, detected both in peripheral plasma and in bone marrow, and severe peripheral vascular involvement. However, we did not find any correlation between EPC apoptotic phenomena and SSc clinical data. This observation could be explained by the fact that AECA recognize antigens shared in mature endothelial cells and endothelial progenitors. In other words, our findings suggest that the deleterious effects of AECA could be the result of their action on both bone marrow and peripheral vascular structures.
Our study demonstrates that antiendothelial activity can also be present in bone marrow plasma from patients with SSc and that it is able to locally induce an increased apoptosis of EPC, in a way similar to what has been observed in the peripheral vasculature. That the purified IgG fractions from the bone marrow plasma reproduce this phenomenon in vitro on CD133+ cells from healthy subjects suggests that this autoantibody activity is directed against the normally expressed surface antigens of these cells.
Our results indicate that an additional AECA-mediated mechanism could be operative in defective vasculogenesis in SSc by inducing apoptosis of EPC in bone marrow. It will be interesting to study the mechanisms involved in AECA-induced endothelial apoptosis further, with the aim of identifying possible inhibitors of the apoptotic pathways.
Acknowledgment
We thank Dr. Daniele Fanoni for his assistance with the immunofluorescent photography and Wendy Doherty for English revision of the manuscript.
- Accepted for publication May 3, 2010.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}