

Abstract
Objective. Pulmonary arterial hypertension (PAH) remains challenging to treat, especially in association with scleroderma. We examined survival rates among patients with PAH in association with scleroderma who received epoprostenol (Flolan®) through continuous intravenous (IV) infusion in an uncontrolled open-label 3-year extension study following an initial randomized, controlled 12-week study.
Methods. One hundred two patients diagnosed with PAH in association with scleroderma who received epoprostenol were included in the analyses. This included 51 PAH patients from a subject population of 56 who received epoprostenol in the randomized controlled study, and 46 patients from an initial population of 55 subjects on conventional therapy in the randomized controlled study, who received epoprostenol in the extension study. All patients in this extension study received open-label epoprostenol. Adverse events, survival, and dosing information were collected throughout the study.
Results. The probabilities of survival during the first and second years for all subjects who received epoprostenol during the initial randomized controlled study or during the extension study were 0.71 and 0.52, respectively. This measure remained constant at 0.48 during the third and fourth years.
Conclusion. This study reports longterm survival rates for patients with scleroderma-associated PAH treated with IV epoprostenol. Although comparisons to historical data should be made with caution, this study reports a better survival outcome than natural history data on patients with scleroderma-associated PAH.
The World Health Organization (WHO) has classified pulmonary hypertension into 5 groups. Group I encompasses a group of diseases, including pulmonary arterial hypertension (PAH) associated with connective tissue vascular disease (i.e., scleroderma). In these diseases, remodeling of the small pulmonary arteries occurs due to histopathological changes that include fibrosis, increased endothelial and medial thickness, and plexiform lesions. PAH is characterized by a sustained increase in the mean pulmonary arterial pressure to > 25 mm Hg at rest, with a mean pulmonary-capillary wedge pressure and/or left ventricular end-diastolic pressure < 15 mm Hg. The disorder is defined further by a progressive increase in pulmonary vascular resistance, culminating in right ventricular failure and eventually death, often by right-heart failure1–5.
Patients with scleroderma are at a higher risk of developing PAH. A recent report from a German study group revealed a 14% occurrence rate of the disease within the scleroderma population, which fits estimates from international data of 12% to 27%6. Other reports expand this prevalence to a range of 4% to 38%7–11. PAH is recognized as a significant clinical component of the scleroderma spectrum of disease8,12,13, which encompasses systemic sclerosis, including diffuse and limited cutaneous variants, mixed connective tissue disease, and overlap syndrome14. Other estimates place the prevalence of PAH at roughly 33% of diffuse cutaneous variants of systemic sclerosis12, reaching up to 50% in limited scleroderma8,13. While numerous disorders are observed in the spectrum of disease, including chronic aspiration, airway disease, neuromuscular weakness, and pleural effusions, the most common causes of death are PAH and interstitial lung disease15,16. Moreover, patients with PAH occurring in association with scleroderma have a poorer outcome following therapy, and a lower longterm survival rate than patients with idiopathic PAH17.
We describe longterm survival data in patients who received chronic intravenous (IV) epoprostenol (Flolan®), either in an initial randomized controlled 12-week study (third-party blind, i.e., defined as site staff performing the 6-minute walk distance (6-MWD) test were blinded to the patients’ treatment arm) or in an uncontrolled open-label 3-year extension study designed to assess longterm safety and survival in patients with moderate to severe PAH occurring in association with scleroderma.
MATERIALS AND METHODS
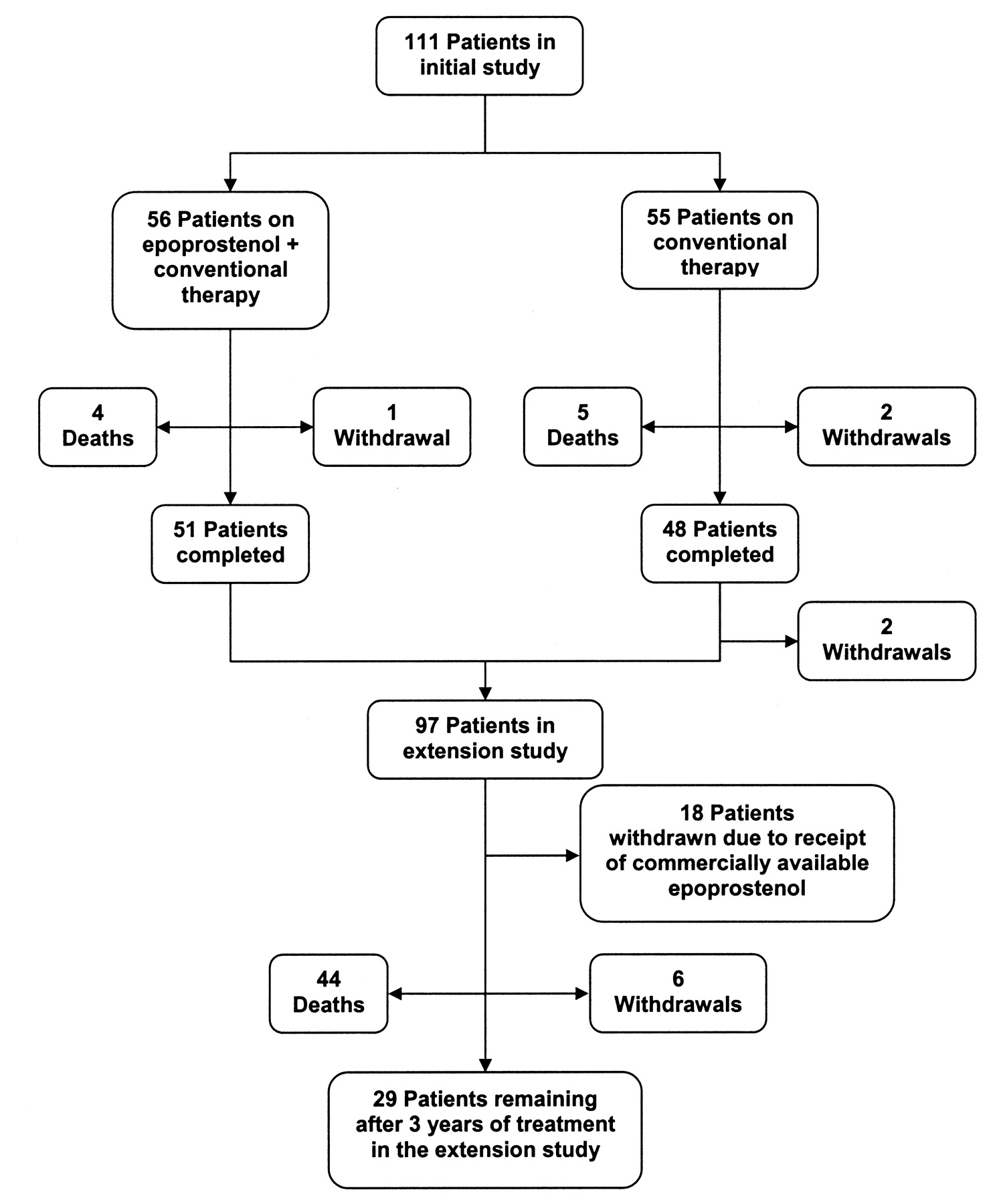
The initial trial, a North America-based multicenter, randomized controlled study of a comparison of continuous IV epoprostenol infusion plus conventional therapy to conventional therapy alone, included 111 study subjects with functional class III or IV PAH occurring in association with scleroderma18, with 56 patients in the former treatment group and 55 patients in the latter. For inclusion in the initial trial, patients must have had a diagnosis of systemic sclerosis, limited scleroderma, overlap syndrome, or definite features of the scleroderma spectrum of disease, and moderate to severe pulmonary hypertension, a ventilation/perfusion lung scan or pulmonary angiography not indicative of thromboembolic disease, and pulmonary function tests and/or high resolution computed tomography scanning showing not more than mild interstitial lung disease. During the study, epoprostenol was administered continuously through a central venous catheter with an ambulatory, positive-pressure pump (CADD-1 HFX model 5100; Sims Deltec, Inc., Saint Paul, MN, USA). Ninety-nine patients with PAH from the initial study were eligible for the extension study, 97 of whom consented to participate in the extension study (Figure 1). This study population included the 51 remaining patients from the epoprostenol plus conventional therapy treatment and 46 patients undergoing conventional therapy, who consented to participate in the extension study. The outcome measures of the initial study were exercise capacity as assessed by the 6-MWD, cardiopulmonary hemodynamics, signs and symptoms of PAH and scleroderma, and survival. Significant treatment effects were seen for 6-MWD and cardiopulmonary hemodynamics, but there was no apparent effect on survival over the course of the 12-week study. However, it was known in advance that the study was not adequately powered to detect an effect on this secondary outcome measure. The majority (93%) of the patients in the initial trial were receiving cardiovascular medications at baseline, with 55% of patients in both treatment groups receiving furosemide. The next most prescribed cardiovascular medications were warfarin, digoxin, and nifedipine, used by 49%, 29%, and 22%, respectively, of patients randomized to conventional therapy, and 36%, 29%, and 29%, respectively, of patients in the epoprostenol group. More patients in the epoprostenol group (86%) than in the conventional therapy group (67%) were receiving warfarin. The study protocol recommended that patients in the study be maintained on anticoagulant therapy unless a contraindication developed. Oxygen use was constant from baseline through the study, at 68% in the epoprostenol group and 76% in the conventional therapy group.

The progress of patients through the study.
The extension companion study was an open-label, uncontrolled study, designed to include all consenting subjects from the previous randomized controlled study to either continue epoprostenol therapy, for those subjects initially randomized to epoprostenol, or start epoprostenol therapy for counterpart subjects earlier randomized to conventional therapy. Eligible patients for this extension study were those who completed all assessments in the earlier study and met all inclusion and exclusion criteria for the study protocol. Prospective subjects for the extension study had to decide whether or not to enroll in this extension study within 1 week following completion of the earlier controlled study. Patients who had been randomized to receive epoprostenol during the earlier controlled trial continued in this extension study at their established infusion rate unless a dosage adjustment was clinically warranted. Patients who received only conventional therapy during the controlled study were started on continuous infusion of epoprostenol at 2 ng/kg/min, and dosage was increased based on tolerability. Adverse events, survival, and dosing information were collected for all patients throughout the study. Patients were eligible to continue receiving epoprostenol under this protocol until they met termination criteria as shown in Table 1. Epoprostenol was administered via a central venous catheter by ambulatory infusion pump. Study subjects were maintained on anticoagulant therapy sufficient to maintain an international normalized ratio between 1.5 and 2.0, unless otherwise contraindicated. All other medicines were allowed as deemed necessary.
List of criteria for patient termination from the extension study.
Ninety-seven patients diagnosed with PAH associated with scleroderma that received epoprostenol are included in the analyses. This population of patients with scleroderma spectrum of disease encompasses systemic sclerosis with diffuse cutaneous and limited cutaneous variants, and mixed connective tissue disease. Patients enrolled in the extension open-label study are those study subjects that survived through the initial study while receiving epoprostenol and those on conventional therapy, which could constitute a potential selection bias.
Analyses were based on time since first receiving epoprostenol. The survival curves for all patients were calculated using Kaplan Meier product-limit estimates. Patients who did not die were censored at the time of withdrawal from either study or study discontinuation. For a limited reference comparison, survival for historical controls was approximated using published data from Koh, et al19.
RESULTS
The current uncontrolled open-label extension study included a population of 102 (Figure 1) eligible patients with functional class III or IV PAH associated with scleroderma (Table 2), 97 of whom were enrolled. Demographically, the study population comprised 90% women, 86% Caucasian, with a mean age of 55 years, and 79% fitting the New York Heart Association Class III at baseline, with 71% of patients having been diagnosed with limited scleroderma. The median duration of PAH was 8 months and the median duration of scleroderma was 57 months within the study population. These figures were comparable to the 17 patients from Koh, et al, consisting of 88% women, 94% Caucasian, mean age 52.5 years, 65% of whom had a diagnosis of limited scleroderma, and a mean duration of scleroderma of 5.5 years19. Within the enrolled subject population, 37 (36%) subjects were diagnosed with PAH within 6 months or less of first receiving epoprostenol. Twenty-four (23%), 23 (23%), 11 (11%), and 7 (7%) subjects were diagnosed between 1 year to 6 months, 1 to 2 years, 2 to 5 years, and more than 5 years, respectively, prior to receiving epoprostenol treatment (Table 3).
Patient demographics and characteristics in the extension study per survival outcome.
Time from diagnosis of pulmonary arterial hypertension to epoprostenol treatment for all subjects enrolled in the study.
From the population of 37 enrolled subjects with a PAH diagnosis within the 6 months prior to receiving epoprostenol, the probabilities of survival during the first, second, and third year post-PAH diagnosis were 0.66, 0.49, and 0.45, respectively (Table 4). The limited number of patients remaining in the study during the fourth year did not allow meaningful data analysis for that timeframe. Of the 61 enrolled subjects with a PAH diagnosis within 1 year prior to receiving epoprostenol, the probabilities of survival within the first and second years post-diagnosis were 0.67 and 0.46, respectively. These measures remained stable, at 0.44, during the third and fourth years following diagnosis. Eighty-four subjects had a PAH diagnosis within 2 years prior to undergoing epoprostenol treatment, and the probability of survival for these patients during the first year post-diagnosis was 0.72 and 0.49 during the second year. These values remained constant at 0.47 during the fourth and fifth years. Upon consideration of the total population of 102 PAH patients eligible, 97 were enrolled to undergo epoprostenol treatment in this extension study. In the latter group 29 patients remained alive after 3 years and 44 deaths occurred. Of 102 total, 28, 17, and 3 patients died during the first, second, and third years, respectively. The probabilities of survival during the first and second years post-diagnosis were 0.71 and 0.52, respectively. These measures remained constant at 0.48 during the third and fourth years (Figure 2). The most common cause of death was right-heart failure, involved in 21 (22%) deaths. The remaining deaths were subsequent to various adverse events (Table 5). None of the deaths was judged by the investigators to be related to epoprostenol or the drug delivery system, and most were attributable to the progression of the disease.

Survival probabilities of patients receiving epoprostenol. Survival rate: the percentage of subjects from the total population alive at the start of the year interval. Patients at risk: the number of subjects assessed at the start of the year interval. Deaths: the number of subjects who died during the following year interval. Censored: the number of subjects who dropped out of the study (lost to followup) during the following year interval.
Survival of subjects receiving epoprostenol following a diagnosis of PAH within a period of 6 months or less.
Summary of adverse events with a fatal outcome.
Four patients, 3 of whom were on conventional therapy and one on epoprostenol during the initial trial, withdrew consent to participate and were withdrawn from the extension study. No specific reason for withdrawal was given by 3 patients, while the fourth noted jaw pain and diarrhea as too uncomfortable to tolerate. Eighteen patients were withdrawn from the study as a consequence of their receipt of commercially available epoprostenol, as required per study protocol.
All 97 patients enrolled in the extension study reported at least one adverse event. The most common adverse events reported (by ≥ 25% of patients) during the extension study were diarrhea, jaw pain, nausea, headache, pain, rash, flushing, depression, right-heart failure, and infection. Sixty-six patients reported at least one serious adverse event and 22 patients experienced right-heart failure, diagnosed based on patients’ history of hospitalizations for paracentesis for massive ascites, IV diuresis and elevated jugular venous pressure, symptomatology, and investigators’ opinion. During the study, common adverse events attributed to the IV delivery system included catheter-mediated sepsis and skin reaction and hemorrhage at the injection site. Thirty patients experienced at least one event leading either to decreases in the epoprostenol infusion rate or to temporary discontinuation of epoprostenol. Two patients, formerly on conventional therapy, withdrew from the study due to adverse events, including one patient who experienced respiratory distress on the first day of epoprostenol infusion necessitating permanent discontinuation of the medication, and she died 5 days later. The other patient became hypotensive, necessitating a decrease in dosing; there was no improvement in blood pressure and the patient was withdrawn permanently from the study. Data from all 97 patients were included in the safety statistical analysis.
DISCUSSION
In this analysis of patients with PAH occurring in association with scleroderma participating in a multicenter uncontrolled open-label extension study of IV epoprostenol, we report survival and serious adverse events, and provide a limited comparison to historical data. Taking into consideration the total eligible subject population of 102 patients regardless of time of PAH diagnosis, 73% of the subjects who underwent epoprostenol treatment survived past the first year. These data indicate, generally, a better outcome in comparison to the report from Koh, et al19 that revealed an extremely poor prognosis of approximately 50% survival rate at 1 year following a diagnosis of PAH associated with scleroderma. Several other studies have addressed survival in patients with PAH occurring in association with scleroderma, and their findings support that this is a very challenging disease to treat20–23.
There is a paucity of data in the literature regarding the efficacy of IV epoprostenol treatment in PAH occurring in association with scleroderma. Our findings complement the previously reported 12-week randomized controlled study involving 111 PAH patients with scleroderma18. That initial study recorded significant improvements in the 6-MWD and hemodynamic measures, but no significant difference in survival between treatment arms18. However, it is important to note that the initial study was not adequately powered to detect a survival difference.
A previous 12-week study of 81 patients with severe idiopathic PAH, which compared conventional therapy alone to continuous IV epoprostenol infusion therapy plus conventional therapy, reported improved exercise capacity, cardiopulmonary hemodynamics, and survival24. However, available data appear to indicate a comparatively lower rate of survival in patients with PAH occurring in association with scleroderma receiving treatment with epoprostenol than in patients with idiopathic PAH23,25.
There are some common, widely reported side effects of epoprostenol therapy, including headache, jaw pain, diarrhea, nausea, rash, and musculoskeletal aches and pains18,24,26. However, these observed side effects tend to be dose-dependent, and can often be managed with dose adjustments. Equally important is the likelihood of exacerbation of pulmonary hypertension or even death that can result from an abrupt interruption of the IV infusion.
There are several limitations in our current extension study. Patients had varying time intervals from diagnosis to epoprostenol treatment. These intersubject differences in time from diagnosis to treatment could have had an effect on their disease state and progression, and subsequently on the outcome of the epoprostenol treatment during the 3-year study. Also, per protocol, subjects who received commercially available epoprostenol were withdrawn from the study and not included in the analysis. Moreover, the survivors from the conventional therapy group in the randomized controlled study proceeded to the extension companion study and received epoprostenol treatment for the first time, which could have engendered a subject selection bias comparative to patients that received epoprostenol through the 2 studies. Finally, any attempt to compare these data to historical reports, such as that by Koh, et al19, is severely limited by potential differences in the study populations, methods of data collection, and background therapies, such as anticoagulation with warfarin.
Despite these shortcomings, our study adds to available information on the probability of survival with the use of epoprostenol in PAH associated with scleroderma.
Footnotes
-
Supported by GlaxoSmithKline, Research Triangle Park, NC, USA.
- Accepted for publication May 27, 2009.








{kind=link}
{kind=link}