

Abstract
Objective. To report the rates of serious adverse events (SAE), serious infectious events (SIE), and events of medical interest (EMI) in patients receiving etanercept; to identify the risk factors for SAE, SIE, and EMI; and to report time to switching from etanercept therapy, reasons for switching, and time to restarting treatment with etanercept in patients with rheumatoid arthritis (RA) in US clinical practice.
Methods. Adults ≥ 18 years of age who fulfilled the 1987 American Rheumatism Association criteria for RA were eligible for enrollment in 2 prospective, 5-year, multicenter, observational registries. RADIUS 1 (Rheumatoid Arthritis DMARD Intervention and Utilization Study) enrolled patients with RA who required a change in treatment [either an addition or a switch of a biologic or nonbiologic disease-modifying antirheumatic drug (DMARD)]. In RADIUS 2, patients with RA were required to start etanercept therapy at entry. Patients were seen at a frequency determined by their rheumatologist. RADIUS 1 and RADIUS 2 were registered under the US National Institutes of Health ClinicalTrials.gov identifiers NCT00116714 and NCT00116727, respectively.
Results. In these patients, SAE, SIE, and EMI occurred at rates comparable to those seen in clinical trials. No unexpected safety signals were observed. Rates for SAE, SIE, and EMI in etanercept-treated patients were comparable to rates observed in patients receiving methotrexate monotherapy and did not increase with greater exposure to etanercept therapy.
Conclusion. The RADIUS registries provide a better understanding of the safety of etanercept in patients with RA in the US practice setting.
Randomized, controlled clinical trials (RCT) provide the strongest scientific evidence for the assessment of safety and efficacy of therapeutic agents1. However, RCT have limitations2. These trials evaluate safety and efficacy over a shorter period of time compared to drug exposure in “real-world” clinical practice. The entrance criteria for RCT frequently limit the severity of comorbid diseases, so patient populations in RCT may not be representative of the patient population that will be taking the drug. Data demonstrating efficacy and safety from RCT must therefore be validated in broader patient populations over a longer period3. Patient registries help fulfill the need to determine adverse events and practice patterns in a more diverse “real-world” population of patients with rheumatoid arthritis (RA).
Treatment for RA is not standardized and includes disease-modifying antirheumatic drugs (DMARD) and biologic agents4, the use of which is influenced by many factors, including physician and patient preferences, severity of symptoms, and known tolerability and effectiveness of medications5,6,7. Patients with RA frequently switch from 1 therapeutic agent to another because of lack/loss of efficacy or because of adverse events8,9. One of the biologic agents approved for the treatment of moderately to severely active RA is etanercept10. It is a human soluble tumor necrosis factor (TNF) receptor-Fc fusion protein that binds specifically to TNF and inhibits its interaction with cell-surface TNF receptors.
The Rheumatoid Arthritis DMARD Intervention and Utilization Study (RADIUS) was designed to gain a better understanding of the safety of different treatment patterns prescribed by rheumatologists and the adverse events of DMARD and biologic agents in the US practice setting. The study comprises 2 prospective, 5-year, multicenter, observational registries (RADIUS 1 and RADIUS 2) of over 10,000 patients with RA11,12. The objectives of the analyses of RADIUS data described here were (1) to provide estimates of the rates of serious adverse events (SAE), serious infectious events (SIE), and events of medical interest (EMI) in patients receiving etanercept; (2) to identify the risk factors for SAE, SIE, and EMI; and (3) to report the distribution of time to switching from etanercept therapy, reasons for switching, and time to restarting treatment with etanercept.
MATERIALS AND METHODS
Patients
Adults ≥ 18 years of age who fulfilled the 1987 American Rheumatism Association criteria for RA13 were eligible. In RADIUS 1, patients required a change in treatment (either an addition or a switch) with a biologic or nonbiologic DMARD. Patients were excluded from participation in both registries if they were currently enrolled in a clinical trial with treatments or patient visits imposed by a protocol, nursing or pregnant, or had an active infection. In RADIUS 2, patients were required to initiate etanercept therapy at entry. Patients were excluded from RADIUS 2 if they were currently or previously enrolled in RADIUS 1, had a known allergy to etanercept or any of its components, were currently receiving treatment with etanercept, or had been shown to be intolerant or refractory to etanercept therapy.
Study design
Patients were seen at a frequency determined by their rheumatologist. During these visits, investigators performed and recorded routine clinical assessments, including a determination of severity of disease (mild, moderate, or severe) per their standard practice. Additional assessments collected for the RADIUS databases included the Health Assessment Questionnaire-Disability Index (HAQ-DI; self-reported on a scale of 0 to 3), swollen joint count, tender joint count, pain visual analog scale, Physician Global Assessment, Patient Global Assessment, and Clinical Disease Activity Index (CDAI)14. Except for the requirement for a patient to receive etanercept at baseline in RADIUS 2, any biologic or nonbiologic DMARD could be initiated, discontinued, or resumed at the discretion of the investigator throughout the study for both RADIUS 1 and RADIUS 2. RADIUS 1 and RADIUS 2 were registered under the US National Institutes of Health ClinicalTrials.gov identifiers NCT00116714 and NCT00116727, respectively.
Safety events
Safety events collected included SAE, SIE, and EMI. An SAE was defined as any event that was life-threatening, resulted in permanent significant disability or incapacity, required inpatient hospitalization or prolongation of existing hospitalization, induced congenital anomalies or birth defects, or resulted in death. An SIE was defined as an SAE involving infection. EMI included cardiovascular events, malignancies, opportunistic infections, tuberculosis, demyelination, and other adverse events, including both nonserious and serious adverse events. Safety events were coded using the Medical Dictionary for Regulatory Activities (MedDRA). EMI and subcategories of SIE were identified using a predefined search of the MedDRA terms; events were reviewed and confirmed for inclusion in the SIE and EMI subgroups by Amgen Global Safety. The occurrence of an adverse event was attributed to the treatment that a patient was taking at the time of onset.
Statistical analyses
Exposure-adjusted incidence rates for SAE were calculated by dividing the number of events by patient-years of exposure multiplied by 100 to represent events per 100 patient-years; 95% CI were calculated based on a Poisson distribution. Standardized incidence ratios (SIR) were calculated for Surveillance, Epidemiology, and End Results Program (SEER) malignancies. The total exposure to etanercept was calculated for each combination of age and sex groups, and then the expected number of malignancy cases was calculated by age and sex category on the basis of the general population SEER rates. The SIR was subsequently calculated as the observed number of cases in this analysis divided by the expected number of cases. To assess the factors associated with SAE, the zero-inflated Poisson model was employed to accommodate count data with an excessive number of zeros. Covariates assessed included age, gender, race, number of comorbid conditions and individual comorbid conditions, disease severity (using CDAI score prior to onset of an adverse event), disease duration, presence of rheumatoid factor, use of prednisone, and score from the HAQ-DI. The distribution of time to etanercept switch and time to restarting etanercept were estimated using the Kaplan-Meier method.
RESULTS
Patient population
A total of 6185 patients initiated treatment with etanercept during the studies. Most patients were women (79% in RADIUS 1 and 77% in RADIUS 2) and most were white (83% in RADIUS 1 and 81% in RADIUS 2; Table 1). Most patients had rheumatoid factor (73% in RADIUS 1 and 67% in RADIUS 2), and most patients had moderate to severe disease as determined by the investigator. The mean (SD) CDAI scores were 37.5 (16.2) and 36.9 (16.0) in RADIUS 1 and RADIUS 2, respectively. About half (57% in RADIUS 1 and 52% in RADIUS 2) were receiving prednisone at the time of entry.
Baseline demographics and disease characteristics of patients who initiated etanercept therapy at any time during the study.
The majority of patients (82% in RADIUS 1 and 91% in RADIUS 2) had received prior nonbiologic DMARD therapy; of these patients, the mean (SD) number of therapies was 2.3 (1.4) in RADIUS 1 and 2.1 (1.1) in RADIUS 2. Exposure to prior biologic therapies had occurred in 34% of patients in RADIUS 1 and 20% of patients in RADIUS 2; of these patients, the mean (SD) number of biologic therapies was 1.1 (0.4) and 1.1 (0.3) in patients in RADIUS 1 and RADIUS 2, respectively.
At the time of study entry, 6% and 29% of patients in RADIUS 1 and RADIUS 2, respectively, were receiving etanercept monotherapy; 18% and 52% were receiving etanercept plus methotrexate (MTX); 12% and 18% were receiving etanercept plus another DMARD; and 13% and 0% were receiving MTX monotherapy. The remaining etanercept-treated patients included in this analysis initiated etanercept therapy after study entry into RADIUS 1.
The number of patients with followup at Years 1, 2, 3, 4, and 5 were 1021, 947, 845, 737, and 622, respectively, in RADIUS 1; and 4399, 3784, 3241, 2615, and 2159 in RADIUS 2. Over the course of the study, 55 patients (5%) in RADIUS 1 and 273 patients (5%) in RADIUS 2 were lost to followup.
Adverse events in all patients
SAE occurred in 14.7% of patients in RADIUS 1 and in 12.3% of patients in RADIUS 2 (Table 2). Rates of SIE were low (approximately 3%) in both RADIUS 1 and RADIUS 2. Rates of malignancies and serious cardiovascular events were about 1%. Of all enrolled patients, 202 in RADIUS 1 and 142 in RADIUS 2 have died, for adjusted rates of 1.25 and 0.83 events/100 patient-years and standardized mortality ratios (observed vs expected deaths) of 0.88 (95% CI 0.77, 1.01) and 0.81 (95% CI 0.68, 0.95), respectively. The most common SAE occurring at an adjusted rate ≥ 0.25% (exposure-adjusted rates as events/100 patient-years in RADIUS 1; RADIUS 2) were pneumonia (0.84; 0.64), congestive cardiac failure (0.41; 0.28), chest pain (0.33; 0.27), myocardial infarction (0.30; 0.25), atrial fibrillation (0.25; 0.13), chronic obstructive pulmonary disease (COPD; 0.25; 0.16), and cellulitis (0.25; 0.26).
Summary of rates of adverse events across all treatment groups. Rates are calculated as number of events/patient-years × 100 = events per 100 patient-years.
Adverse events in patients receiving etanercept
The relative risk of most events in patients receiving etanercept was comparable to the risk of the event in patients receiving MTX monotherapy (Table 3). Rates of adverse events did not increase with longer exposure to etanercept (Table 4). Rates of SIE over time were similar in patients receiving etanercept as either first-line or second-line biologic DMARD therapy (Table 4). Results for patients receiving etanercept as second-line therapy should be interpreted with caution, however, as the sample sizes were extremely small.
Exposure-adjusted rates of adverse events in all enrolled patients.
Exposure-adjusted rates of adverse events in patients receiving etanercept. Exposure represents time on etanercept. Many patients experienced a gap in etanercept exposure during which other biologics were introduced and discontinued. Event rates were calculated as no. of events/patient-years × 100. Seven patients were not included in the RADIUS 2 analysis because of missing etanercept treatment information. Serious cardiovascular (CV) events include chronic heart failure, coronary artery disease, myocardial infarction, cardiomyopathy, and stroke.
Opportunistic infections in all patients
Twenty-seven opportunistic infections were reported in RADIUS 1 and 31 were reported in RADIUS 2. Two cases of tuberculosis were reported in RADIUS 1 and 3 cases in RADIUS 2. There were 2 cases of disseminated histoplasmosis in RADIUS 1 and 1 case in RADIUS 2.
Demyelination/neurological events in all patients
There was 1 report of demyelination in a patient receiving etanercept in RADIUS 2. The abnormal magnetic resonance imaging findings resolved with discontinuation of therapy. There were 10 and 16 nervous disorder events reported in RADIUS 1 and RADIUS 2, respectively.
Malignancies in all patients
The SIR for SEER malignancies for patients in the full analysis set were 0.81 (95% CI 0.67, 0.95) in RADIUS 1 and 0.86 (95% CI 0.72, 1.03) in RADIUS 2. There were 5 events of melanoma of the skin reported in RADIUS 1 for an adjusted rate of 0.03 events per 100 patient-years (95% CI 0.01, 0.07) and a SIR of 1.00 (95% CI 0.32, 2.33). Four patients were receiving only DMARD therapies and 1 patient was receiving etanercept therapy. There were 8 events of melanoma of the skin reported in RADIUS 2 for an adjusted rate of 0.05 events per 100 patient-years (95% CI 0.02, 0.09) and an SIR of 1.68 (95% CI 0.73, 3.31). All patients were receiving anti-TNF therapy including etanercept. There were 12 events of lymphoma reported in RADIUS 1 for an adjusted rate of 0.07 events per 100 patient-years (95% CI 0.04, 0.13) and an SIR of 1.79 (95% CI 0.92, 3.13). There were 12 events of lymphoma reported in RADIUS 2 for an adjusted rate of 0.07 events per 100 patient-years (95% CI 0.04, 0.12) and an SIR of 2.06 (95% CI 1.06, 3.60).
Risk factor analyses
Risk factors for SAE in either study included age at baseline, number of comorbid conditions at baseline, HAQ index at prior visit or at each treatment segment, and prednisone use at the start of each treatment segment (Table 5). In RADIUS 1, the odds of having an SAE increased by 134% for each additional comorbid condition and increased by 58% for each unit-increase in HAQ score. Also in RADIUS 1, for 2 patients with a 10-year difference in age at baseline, the number of SAE was 1.35 times greater for the older patient than the number expected for the younger patient. In RADIUS 2, the odds of having an SAE increased by 203% for each comorbid condition at baseline, by 7% for each additional year of age, and by 84% for patients using prednisone at the start of each treatment segment. Also in RADIUS 2, the number of SAE would be 1.45 times higher for each unit-increase in HAQ.
Risk factors for adverse events in patients receiving etanercept.
Risk factors for SIE in either study included COPD at baseline, HAQ index at prior visit or at each treatment segment, age, number of comorbidities at baseline, and prednisone use at the start of each treatment segment. In RADIUS 1, the hazard rate for SIE increased by 331% for patients with COPD compared with patients without COPD and increased by 63% for each unit-increase in HAQ score. In RADIUS 2, the hazard rate for SIE increased by 3% for each additional year of age, by 40% for each additional comorbidity, by 30% for each unit-increase in HAQ score, and by 46% for patients using prednisone at the start of each treatment segment.
Risk factors for serious cardiovascular events in either study included number of comorbid conditions at baseline, male sex, age, and comorbid diabetes at baseline. In RADIUS 1, the hazard rate for a serious cardiovascular event decreased by 69% for women compared with men, and increased by 131% for each additional comorbidity at baseline. In RADIUS 2, the hazard rate for a serious cardiovascular event increased by 7% for each additional year of age and by 70% for each additional comorbidity at baseline, decreased by 43% for women compared with men, and increased by 121% for patients with diabetes at baseline.
Covariates of race, disease severity, disease duration, and presence of rheumatoid factor were not found to be risk factors for SAE, SIE, or serious cardiovascular events in this analysis.
Switching from and reinitiating etanercept therapy
Many patients switched therapies over the study period. Reasons for switching from etanercept therapy were lack of efficacy [n = 150 (36%) in RADIUS 1; n = 666 (31%) in RADIUS 2], adverse events [n = 92 (22%); n = 518 (24%)], patient decision [n = 67 (16%); n = 344 (16%)], cost [n = 38 (9%); n = 397 (19%)], and other reasons [n = 64 (15%); n = 189 (9%)]. The distribution of times to etanercept discontinuation and restart are shown in Figure 1. The median time to discontinuation from etanercept was 51.8 months (95% CI 44.1, not estimable) in RADIUS 1 and 44.7 (95% CI 42.5, 48.8) months in RADIUS 2.
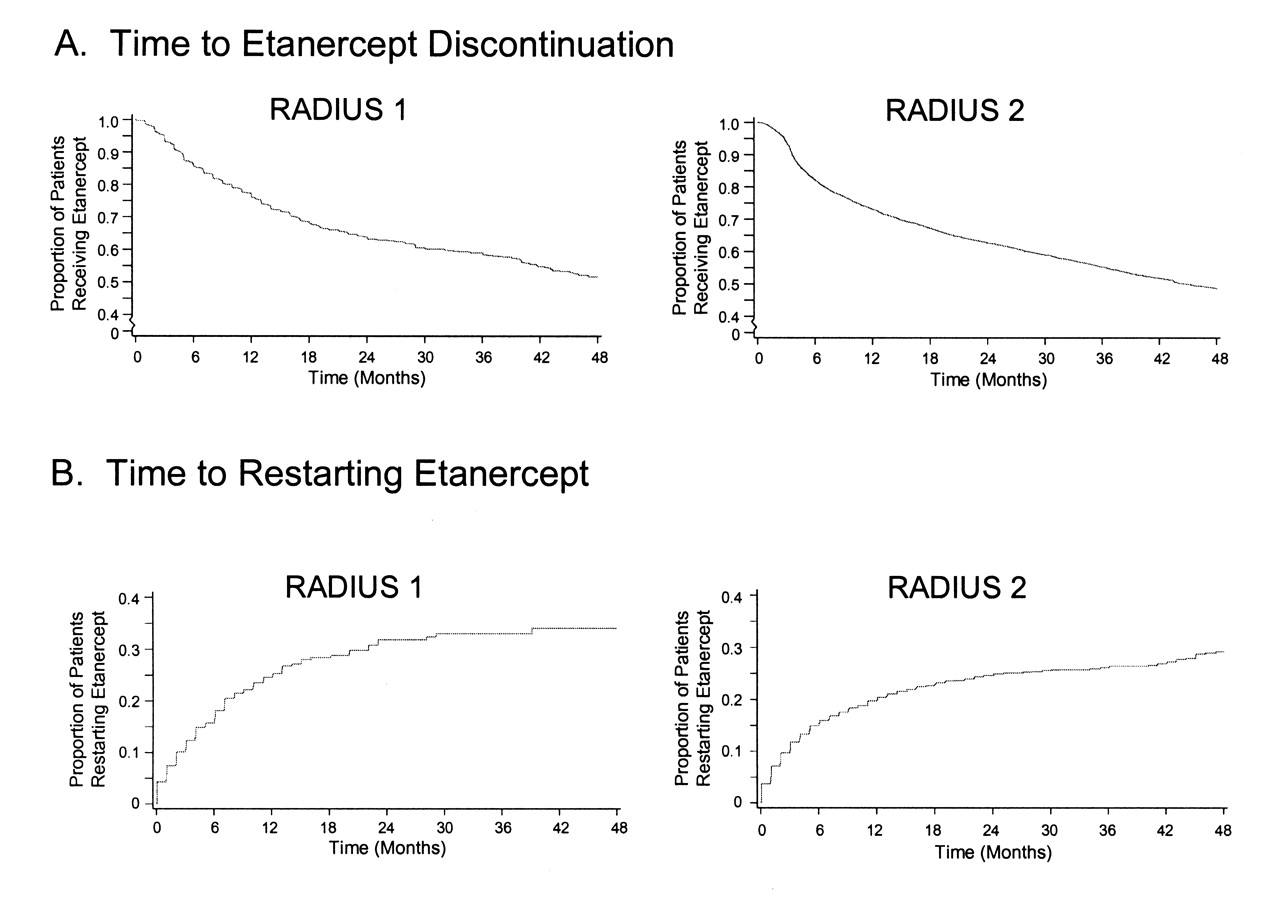
Time to etanercept discontinuation and restart.
DISCUSSION
In these patients with RA enrolled in the RADIUS registries, SAE, SIE, and EMI occurred at rates comparable to those seen in clinical trials. No unexpected safety signals were observed. Patients treated with TNF inhibitors, including etanercept, had decreased rates of death compared to patients receiving DMARD therapies. COPD emerged as a significant risk factor for SIE in both RADIUS 1 and RADIUS 2.
Many patients switched therapies over the study period. The most common reason that patients switched from etanercept therapy was lack of efficacy, which occurred in about one-third of all patients in the study. This rate of switching from a TNF inhibitor is consistent with other studies9. Adverse events leading to a switch from etanercept occurred in about one-quarter of all patients.
Registries provide information on broader patient populations than are seen in RCT. The RADIUS registries have enrolled more than 10,000 patients, providing information on far greater numbers of patients than have been enrolled in RCT. Registries also provide data on longterm use of drugs, increasing the possibility to detect infrequent safety events. Of note, safety data from the RADIUS registries have not identified any unexpected adverse events.
Registries also have limitations, including selection bias and ascertainment bias. Selection bias may occur in RA registries because patients whose disease is adequately controlled may not be enrolled. This may lead to greater representation of patients with long-lasting, severe disease who have had an inadequate response to other DMARD, as well as recently diagnosed patients with RA who are initiating their first DMARD in the RADIUS registries, since patients were required to initiate a new therapy at study entry. Ascertainment bias may occur because the frequency of observations is determined by the individual rheumatologist rather than at prespecified intervals, as in RCT. Some patients may therefore have a greater opportunity to have specific outcomes documented, including adverse events.
Rates of SIE in patients receiving etanercept were comparable to rates in patients receiving MTX in RADIUS. Data from other registries have shown a modest increase for risk of infections with TNF antagonists but self-reported rates are inconsistent. Data from the Consortium of Rheumatology Researchers of North America database (CORRONA; n = 5596) showed an incidence rate ratio of infections (severity not specified) in patients receiving anti-TNF therapies to patients not receiving anti-TNF therapies of 1.16 (95% CI 1.06, 1.28) and an incidence rate ratio for opportunistic infections of 1.4615. Similarly, the Dutch Rheumatoid Arthritis Monitoring Register (DREAM) reported a rate ratio for serious infections of 1.68 (95% CI 1.23, 2.3) in patients receiving anti-TNF therapies compared with patients receiving DMARD16. However, data from the British Society for Rheumatology Biologics Registry (BSRBR; n > 8000) showed no increase in the incidence of serious infections in patients receiving anti-TNF therapies17,18. Similar to our results, HAQ score was identified as a risk factor for infection in CORRONA19, but no other risk factors were common in our database and CORRONA. Our data showed that rates of SIE did not increase with time taking etanercept. Similarly, the relative risk of infections leading to hospitalization in patients receiving anti-TNF therapies in the Anti-Rheumatic Therapies in Sweden registry (ARTIS; n = 44,946) decreased over time.
Our data suggest that the risk of malignancy in patients receiving etanercept is not greater than the risk of malignancy in patients receiving the DMARD MTX. This observation is consistent with data from other registries of patients with RA receiving DMARD and anti-TNF therapies. The relative risk of malignancy in patients receiving anti-TNF therapies was 0.94 (95% CI 0.80, 1.12) in ARTIS (n = 66,995)20. Incidence rate ratios of malignancies in patients receiving anti-TNF therapies compared to patients receiving DMARD have been reported to be 0.7 (95% CI 0.4, 1.2) in the BSRBR (n = 11,875)21; and 0.73 (95% CI 0.51, 1.06) in CORRONA (n = 9123)22. A case-control study of the German Biologics Register Rheumatoid Arthritis Observation of Biologic Therapy (RABBIT; n = 5000) also showed no difference in the incidence of solid malignancies in patients who were exposed or not exposed to biologic therapies23. Treatment with etanercept or infliximab was not associated with an increased risk of cancer overall in patients with RA in the South Swedish Arthritis Treatment Group (SSATG; n = 757)24.
Multiple studies have indicated that incidence of lymphoma is about double in patients with RA compared with the general population25. The SIR for lymphoma were 1.79 and 2.06 in RADIUS 1 and RADIUS 2, respectively, consistent with these other studies. While some studies have suggested that anti-TNF therapies do not further increase the risk of lymphoma in patients with RA25, this question remains unanswered and few registries have addressed this question. Data from the SSATG registry reported a SIR for lymphoma of 11.5 (95% CI 3.7, 26.9) in patients treated with anti-TNF therapies and an overall risk of cancer excluding lymphoma of 0.79 (95% CI 0.4, 1.42)24. A case-control study of data from Recherche Axée sur la Tolérance des Biothérapies (RATIO) suggested that the risk of lymphoma may be higher with adalimumab or infliximab than with etanercept26.
The RADIUS registries provide a better understanding of the safety of etanercept in patients with RA in real-world US clinical practice. Data from RADIUS have shown that the rates for SAE, SIE, and EMI in etanercept-treated patients were comparable to rates observed in patients receiving MTX monotherapy in the study, and did not increase with greater exposure to etanercept therapy. Notably, no unexpected safety signals were observed. Results from this registry and others are important in validating data reported in pharmaceutical longterm extension safety studies and thereby provide information for use in rheumatology practice.
Acknowledgment
The authors thank Meera Kodukulla, PhD, of Amgen Inc. and Julia R. Gage, PhD, on behalf of Amgen Inc. for writing assistance.
Footnotes
-
Supported by Immunex, a wholly owned subsidiary of Amgen Inc., and by Wyeth Pharmaceuticals, which was acquired by Pfizer in October 2009.
- Accepted for publication August 11, 2010.








{kind=link}