

To the Editor:
Secondary amyloidosis can develop in a patient with seemingly quiescent giant cell arteritis/polymyalgia rheumatica (GCA/PMR). We describe a patient with clinically quiescent GCA who developed AA amyloidosis of the kidney, resulting in nephrotic syndrome and acute renal failure.
A 79-year-old woman presented to the rheumatology service in December 2009 with new-onset unilateral headache and prominence and tenderness of the left temporal artery. Erythrocyte sedimentation rate (ESR) was 85 mm/h and C-reactive protein (CRP) was 40.9 mg/l. She fulfilled 4 of the 5 American College of Rheumatology (ACR) 1990 criteria for GCA: age > 50 years, new-onset headache, temporal artery abnormality, and ESR > 50 mm/h1. She denied symptoms of PMR but she had anorexia and generalized fatigue that had been persistent for several years. Subsequent investigations for GCA included left-side temporal artery biopsy of 1 cm and magnetic resonance imaging (MRI) of the scalp vessels with contrast. The temporal artery biopsy was negative for evidence of arteritis; however, the MRI showed abnormal enhancement in the superficial temporal artery in the postcontrast images. This finding was felt to be diagnostic of arteritis.
The patient had a 35 pack-years smoking history with moderate chronic obstructive pulmonary disease (COPD) and a history of tuberculosis treated in 1951 with residual right-upper lobe traction bronchiectasis, demonstrable on computerized tomography (CT) scan. She experienced on average 1 infectious exacerbation per year and in the intervening period had no baseline sputum production. She also had hypertension, paroxysmal atrial fibrillation, and hypothyroidism.
She was initially treated for GCA with prednisone 50 mg daily for 3 weeks, with complete resolution of her symptoms, and was slowly tapered to 15 mg daily as maintenance. Her ESR improved to a nadir of 23 mm/h. In May 2010 she had a relapse of her symptoms and was retreated with high-dose prednisone for 2 weeks with prompt resolution. Three weeks later she was admitted to hospital with shortness of breath and increasing peripheral edema, which was treated as a COPD exacerbation. She was managed with prednisone 50 mg daily on slow taper, antibiotics, bronchodilators, and diuretics. Creatinine was 165 μmol/l, with baseline 94 μmol/l in March of 2009. A 24-hour urine protein collection showed nephrotic-range proteinuria of 3.68 g, and angiotensin receptor blocker therapy was added.
She developed new acute renal failure (peak creatinine 282 μmol/l) in July 2010. Serum and urine protein electrophoresis were negative for M-protein. Also negative were antinuclear antibody, rheumatoid factor, cytoplasmic and perinuclear antineutrophilic cytoplasmic antibody, antiglomerular basement membrane antibody, and hepatitis B and C serology. A 2-hour, 75-g glucose tolerance test demonstrated impaired fasting glucose. Renal biopsy demonstrated nodules of acellular eosinophilic material that was periodic acid-Schiff negative, nonargyrophylic on Jones silver stain, green/orange on trichrome stain, and Congo-red positive. It had apple-green appearance on polarized microscopy. Immunofluorescence showed isointense staining for kappa (2+) and lambda (3+). On immunohistochemistry, amyloid A immunostain was strongly positive. Electron microscopy confirmed the findings, which were diagnostic of AA amyloidosis affecting the kidney. Review of the temporal artery biopsy was negative for amyloid. Bone marrow biopsy showed evidence of amyloid with no evidence of abnormal cell infiltrate.
At the time of diagnosis of AA amyloidosis, the patient did not have symptoms of GCA or PMR. Her ESR was 73 mm/h with CRP 35.4 mg/l. She was receiving 5 mg prednisone daily. Colchicine 0.6 mg daily was initiated, but was decreased to 0.3 mg daily due to diarrhea. At 4 months her creatinine had stabilized at 220 μmol/l, with 24-hour urine protein 3.81 g. She died of hospital-acquired pneumonia 2 months later.
AA amyloidosis is caused by extracellular deposition of fibrils composed of fragments of the acute-phase reactant serum amyloid A (SAA) protein2. Deposition can lead to progressive organ dysfunction, particularly of the kidneys3. AA amyloidosis is found in several rheumatological conditions, most notably rheumatoid arthritis (RA)4. GCA and/or PMR have rarely been associated with amyloid5,6,7,8,9,10,11,12. Among patients with RA, AA amyloidosis has been most commonly identified in more severe and poorly controlled disease, and it is theorized that it is the chronically uncontrolled inflammatory medium that results in elevated levels of the SAA protein and subsequent accumulation as amyloid13. This theory has been supported by the association of AA amyloidosis with other non-rheumatic but inflammatory conditions, such as lymphoma and cystic fibrosis4.
We have undertaken a systematic review of the literature to identify previous cases of AA amyloidosis in patients with GCA and/or PMR. We electronically searched the Medline, Embase, and CINAHL® databases, along with hand-searching bibliographies of retrieved articles, and hand-searching abstracts from the last several years from the annual meetings of the Canadian Rheumatological Association, the ACR, the European League Against Rheumatism, and the International Vasculitis and ANCA Workshop. We chose articles using the keywords “giant cell arteritis” or “polymyalgia rheumatica” and “amyloidosis.” The articles were chosen based on the following criteria: (1) cases from case reports, case series, or previous systematic reviews; (2) biopsy or autopsy-confirmed AA amyloid from any tissue; and (3) diagnosis of GCA and/or PMR. Exclusion criteria were amyloid not of the AA type, and a rheumatological condition other than GCA/PMR.
Searches and application of the inclusion and exclusion criteria were performed by 2 independent reviewers and discrepancies were resolved by consensus. Figure 1 depicts the search results. Summaries of the included cases are presented in Table 1.
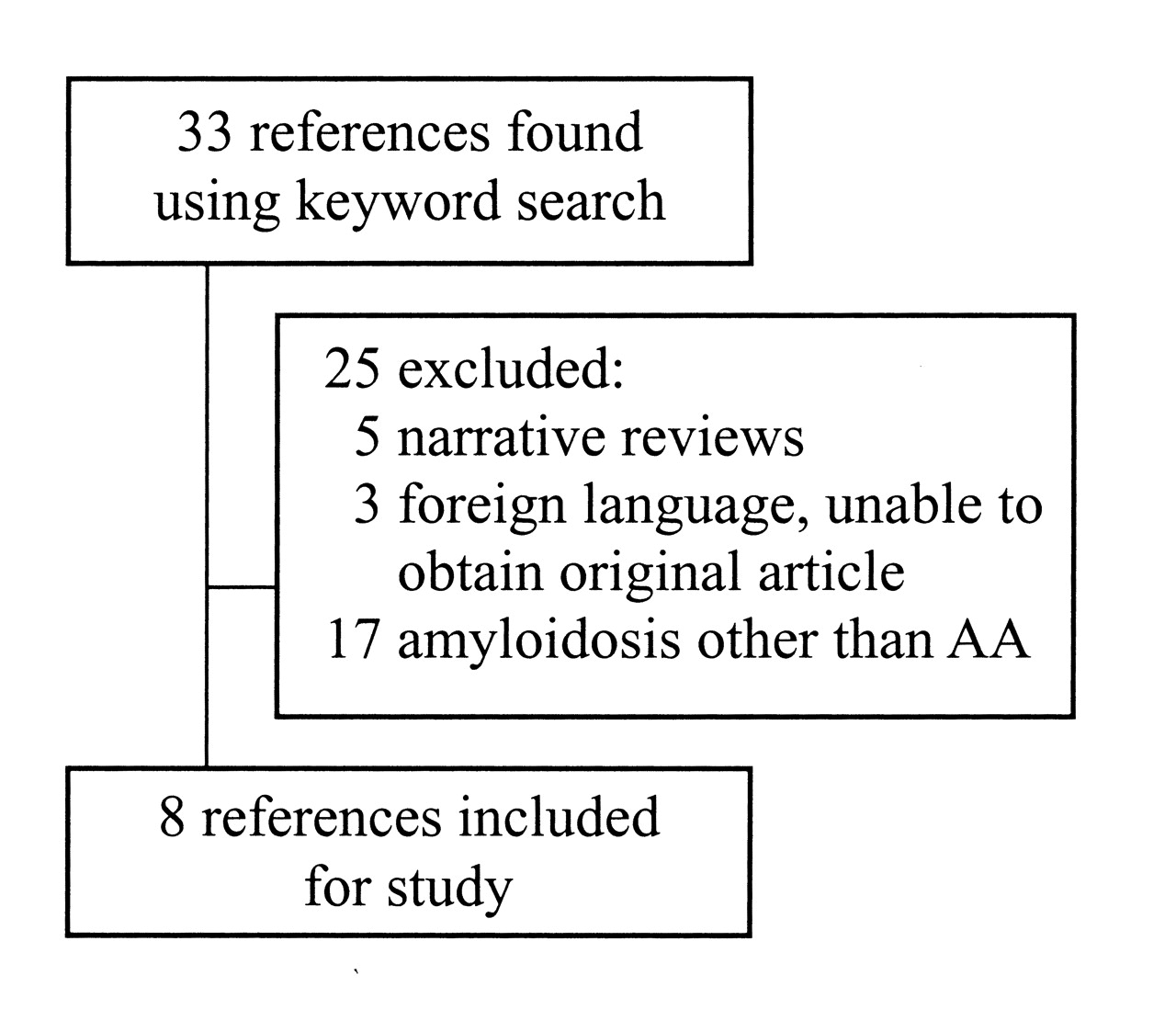
Results of a literature search for reports of AA amyloidosis in patients with giant cell arteritis and/or polymyalgia rheumatica.
Summary of cases of AA amyloidosis in patients with giant cell arteritis and/or polymyalgia rheumatica.
There are several patients described in the series with symptoms suggestive of uncontrolled GCA or PMR for months or years prior to the diagnosis of amyloidosis. Thus it is possible that this secondary amyloidosis is associated, as in RA, with ongoing inflammation5,6,7,10,11. Interestingly, our patient had quiescent disease with normalization of the ESR in the intervening period. She did have evidence of bronchiectasis on CT scan, a condition that has also been associated with AA amyloidosis, but she had been relatively asymptomatic. Two of the previous case reports also described patients with well controlled GCA/PMR who developed AA amyloidosis6,12. This suggests either subclinical disease in seemingly well controlled GCA/PMR, or an alternative mechanism for AA amyloid deposition.
Treatment of AA amyloidosis has historically been treatment of the underlying illness with the aim of decreasing SAA protein production. In our case series, many patients were treated with high-dose steroids. Although 1 patient treated with low-dose prednisone showed stabilization of renal function over 2 years of followup12, renal recovery was not achieved in any of these cases using prednisone alone, and most patients died of progressive renal failure within months to 2 years5,6,11. In familial Mediterranean fever, a condition commonly associated with AA amyloidosis, the incidence of amyloidosis is lower in those patients treated with colchicine, and there have been reports of improvement in proteinuria14 and even reversal of nephrotic syndrome15,16. Our patient, along with the 2 described by Monteagudo, et al9, was treated with colchicine with evidence of stabilization or improvement in renal function. These results are encouraging as colchicine has a good safety profile and is relatively easy to use. Chlorambucil has been used in 2 patients; however, 1 patient progressed to hemodialysis and the outcome of the second is unknown8,10. There is limited or no experience with newer immunosuppressives or biological agents.
Our case raises the possibility of secondary amyloidosis developing in a patient with seemingly quiescent GCA/PMR, which highlights the need for screening with periodic serum creatinine measurement and urinalysis for protein. Colchicine may be effective at preventing or delaying progression of renal failure in AA amyloid.








{kind=link}