

Abstract
Objective. It was previously found that Th1 but not Th17 cells were predominant in the joints of rheumatoid arthritis (RA). To verify whether this is a unique feature of CD4 T cells in RA joints, we performed comparative flow cytometric analysis of CD4 T cells in RA and osteoarthritis (OA) joints.
Methods. Mononuclear cells were isolated from peripheral blood (PB), synovial membrane (SM), and synovial fluid (SF) from a total of 18 RA and 12 OA patients. The expression of surface molecules and cytokine production of CD4 T cells was examined by a flow cytometer.
Results. Most CD4 T cells in RA joints expressed memory/activation markers, such as CD45RO, HLA-DR, and CD69. CCR5 was highly expressed on CD4 T cells in SF but not in PB or SM. With regard to Th17-related molecules, CD4 T cells expressing CCR6 were not enriched in either SF or SM. In contrast, CD161-positive cells were abundant in the joint, many of which, however, produced interferon-γ but not interleukin 17A. Virtually all T cells in OA joints, although much less numerous than in RA joints, expressed activation markers. Th1 cells were predominant in both OA and RA joints, while there were a few Th17 cells. The frequency of Th17 cells in the joint tended to be lower in OA than RA.
Conclusion. There was a quantitative but not qualitative difference in CD4 T cells, including the expression of activation markers and cytokine profiles, between RA and OA joints.
Rheumatoid arthritis (RA) is characterized by chronic and destructive inflammation of systemic joints, which is thought to be driven by autoreactive CD4 T cells. This is suggested by the association of the disease with certain MHC class II haplotypes and by the infiltration of activated CD4 T cells in the inflamed synovium1,2. Recently, identification of protein tyrosine phosphatase, non-receptor type 22 (PTPN22), which is involved in T cell receptor signaling, as a genetic risk factor and the clinical efficacy of blocking the T cell costimulatory molecule CD28 by cytotoxic T-lymphocyte antigen 4 (CTLA4)-Ig add further support to the hypothesis3,4. CD4 T cell-dependent development of various animal models of RA is also an important rationale. Due to their ability to induce delayed-type hypersensitivity reaction, Th1 cells had long been believed to be pathogenic among CD4 T cell subsets. However, such a Th1 paradigm was challenged after the discovery of Th17 cells. Human Th17 cells are a unique subset of helper T cells that produce interleukin 17A (IL-17) and are phenotypically characterized by the expression of C-C chemokine receptor type 6 (CCR6) and CD1615. CCR6 is the receptor for chemokine (C-C motif) ligand 20 (CCL20), while CD161 is the human ortholog of mouse natural killer antigen 1.1 (NK1.1). Lines of evidence suggest the importance of IL-17 in the pathogenesis of RA6. IL-17 has a variety of biological activities that explain well the destructive expression of inflammation in RA. IL-17 is essential for the development of various animal models of arthritis. Elevated levels of IL-17 production in RA joints have been reported. These findings led to the belief that RA is a Th17-mediated autoimmune disease.
However, in a previous study, we unexpectedly found that there was either no increase of Th17 cells in the peripheral blood (PB) of RA or no correlation between the number of Th17 cells and disease activity of RA7. More importantly, it was revealed that Th1 but not Th17 cells predominated in RA joints. Similar data were shown in other studies8,9,10. These findings questioned the Th17 hypothesis of RA. However, it remains possible that Th17 cells in RA joints are refractory to detection by means of intracellular staining of cytokines, as joint-infiltrating T cells are known to be hyporesponsive11. Moreover, it is unclear whether such predominance of Th1 cells is a unique feature of CD4 T cells in RA joints.
Osteoarthritis (OA) is a chronic disorder of the joint affecting a large population worldwide. Although OA joints also show clinical signs of inflammation, i.e., pain, swelling, local heat, and redness, OA is generally regarded as a degenerative disease. The radiographic changes of OA joints are distinct from those of RA in that OA joints tend to show sclerotic changes in the subchondral bone and osteophyte formation. Synovitis, even in a villous form, is occasionally observed in OA joints, but its histology shows much less numerous CD4 T cells and macrophages than RA synovium12. There is also a large difference in the cellularity of synovial fluid. Therefore, although many studies on RA have used OA samples as the negative control and showed an increased production of inflammatory mediators in RA, this could simply reflect the quantitative difference in cellular infiltrates. However, provided that RA but not OA is likely a CD4 T cell-mediated autoimmune disease, there could also be qualitative differences in CD4 T cells in the joints between RA and OA. We first examined expression of activation markers and chemokine receptors, including Th17-associated markers, on CD4 T cells in RA joints, and then compared the phenotypes with those in OA joints to characterize RA CD4 T cells. The Th1/Th17 profile of CD4 T cells in RA and OA joints was also analyzed.
MATERIALS AND METHODS
Patients
Eighteen patients with RA (15 women, 3 men, mean age 61 ± 12 yrs) diagnosed on the 1987 criteria of the American College of Rheumatology13, and 12 patients with OA (8 women, 4 men, mean age 66 ± 11 yrs) were included in the study. The duration of RA ranged from 1 to 34 years (mean 13 ± 10 yrs). Fifteen RA patients were taking disease-modifying antirheumatic drugs (DMARD; 10, methotrexate; 2, bucillamine; 2, tacrolimus; 1, auranofin; 1, leflunomide; 1, gold sodium thiomalate) either as monotherapy or in combination. One patient was treated with infliximab in combination with methotrexate. Nine patients were taking prednisolone (mean 5 mg/day). No patient had received intraarticular steroid within 1 month before analysis. We evaluated radiographic changes of joints using Larsen and Kellgren-Lawrence grading systems for OA and RA, respectively (Table 1).
Patients’ characteristics.
The study protocol was approved by Regional Committee of Ethics for Human Research at the Faculty of Medicine of Kyushu University. All subjects provided signed informed consent before the study.
Preparation of mononuclear cells
Synovial membrane (SM) was removed at the time of joint replacement surgery. Samples were minced and incubated with 2 mg/ml of collagenase (Wako, Osaka, Japan) for 90 min at 37°C. A single cell suspension was layered on Ficoll-Paque Plus separation media (GE Healthcare Bio-Science AB, Uppsala, Sweden) and centrifuged to harvest the mononuclear cells on the interface. PB mononuclear cells were similarly separated using Ficoll-Paque Plus. Synovial fluid (SF) was obtained by arthrocentesis, when the patients claimed symptomatic effusion. Thus, all these samples were obtained from joints with active inflammation. Two SF samples were obtained during surgery. For isolation of SF mononuclear cells, SF was treated with 20 μg/ml hyaluronidase (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37°C and was then centrifuged. The pellet was dissolved in phosphate buffered saline and subjected to Ficoll-Paque Plus density gradient centrifugation.
Flow cytometric analysis
Monoclonal antibodies (mAb) and reagents used for flow cytometric analysis comprised Alexa Fluor 488-conjugated anti-IL-17A mAb (eBio64DEC17; e-Bioscience, San Diego, CA, USA), fluorescein isocyanate-conjugated anti-CD57 (TB01; Caltag Laboratories, Burlingame, CA, USA) and anti-CD45RO (UCHL1; e-Bioscience) mAb, phycoerythrin-conjugated anti-CD28 (15E8; Caltag), anti-CCR6 (11A9; BD Bioscience, San Diego, CA, USA), and anti-interferon-γ (4S.B3; e-Bioscience) mAb, allophycoerythrin-conjugated anti-CD4 (S3.5; Caltag), PerCP-Cy5.5-conjugated anti-CD3 (SK7; BD Bioscience) and anti-CD161 (HP-3G10; e-Bioscience) mAb, biotin-conjugated anti-CD4 (S3.5; Caltag), anti-HLA-DR (LN3), anti-CD69 (FN50), and anti-CCR5 (eBioT21/8; e-Bioscience) mAb, and streptavidin-allophycoerythrin and streptavidin-phycoertythrin (e-Bioscience). Stained cells were run on a FACS Calibur flow cytometer (BD Bioscience), and the data were analyzed using CellQuest software (BD Bioscience).
Intracellular staining of cytokines
Mononuclear cells were stimulated with 50 ng/ml phorbol myristate acetate (PMA; Sigma-Aldrich) and 1 μg/ml ionomycin (Sigma-Aldrich) in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich) for 4 h. After surface-staining of the cells, intracellular staining was performed using BD Cytofix/Cytoperm solution and BD Perm/Wash solution following the manufacturer’s instructions (BD Biosciences).
Statistics
Mann-Whitney U-test was used for statistical analysis of the difference between 2 groups in all experiments. P values less than 0.05 were considered significant.
RESULTS
T cells in RA joints uniformly expressed activation markers
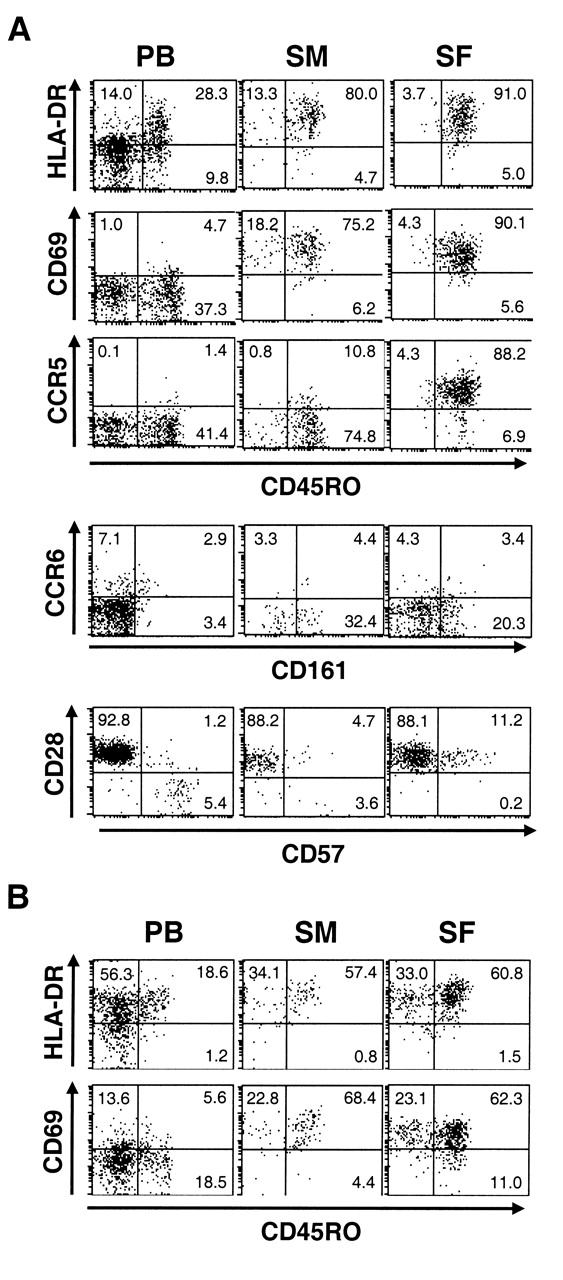
We first compared the phenotypes of CD4 T cells in PB to those in SM and SF of patients with RA (Figure 1A). Most CD4 T cells in the joint expressed a memory T cell marker, CD45RO. The expression levels of activated T cell markers such as HLA-DR and CD69 were higher in CD45RO+ CD4 T cells in the joints than those in PB. Interestingly, CD28-negative CD4 T cells, which express CD57 and are thought to be involved in the pathogenesis of RA14, decreased in both SM and SF compared with PB. Instead, a population of CD28+ cells expressing intermediate levels of CD57 appeared in the joint. When we examined CD4-negative T cells (CD4–CD3+), in which more than 90% were CD8 T cells, we noted that most of those in the joint expressed activation markers (Figure 1B), similar to CD4 T cells. Thus, virtually all T cells in RA joints showed activated T cell phenotypes.

Expression of surface molecules on T cells in RA. Mononuclear cells isolated from peripheral blood (PB), synovial membrane (SM), and synovial fluid (SF) of RA patients were analyzed for expression of surface molecules. Representative dot plots after gating on (A) CD4+ or (B) CD4–CD3+ cells are shown.
Expression of Th17-associated markers on CD4 T cells in RA joints
We next examined the expression of Th17-associated markers. PB CD4 T cells expressing CCR6 were clearly detected in the CD45RO+ memory T cell population (Figure 2A). However, the frequency of CCR6+ cells did not increase in either SF or SM compared with PB. Indeed, the frequency of CCR6+ cells within CD45RO+ cells was even lower in the joint than in PB (Figure 3B). In contrast, CD4 T cells expressing CCR5, which is a marker of Th1 cells15, were more numerous in SF than in PB and SM, as reported previously16,17. CD161 is another marker of Th17 cells, and we found its expression on about 30% of CD45RO+ CD4 T cells in SM, SF, and PB, but not on CD45RO-negative cells. However, as expected, a majority of CD161+ CD4 T cells in the joint were CCR6-negative, but were CCR5-positive (data not shown). Moreover, intracellular staining revealed that Th1 cells were dominant over Th17 cells in CD161+ cells in the joints (Figure 2B). The frequency of Th1 cells increased significantly in CD161+ cells in SF, whereas frequency of Th17 cells in CD161+ was lower in SM and SF than in PB (Figure 2C). These results suggest that an accumulation of unresponsive CD161+ cells seems unlikely to be the explanation for the relative paucity of Th17 cells in RA joints.
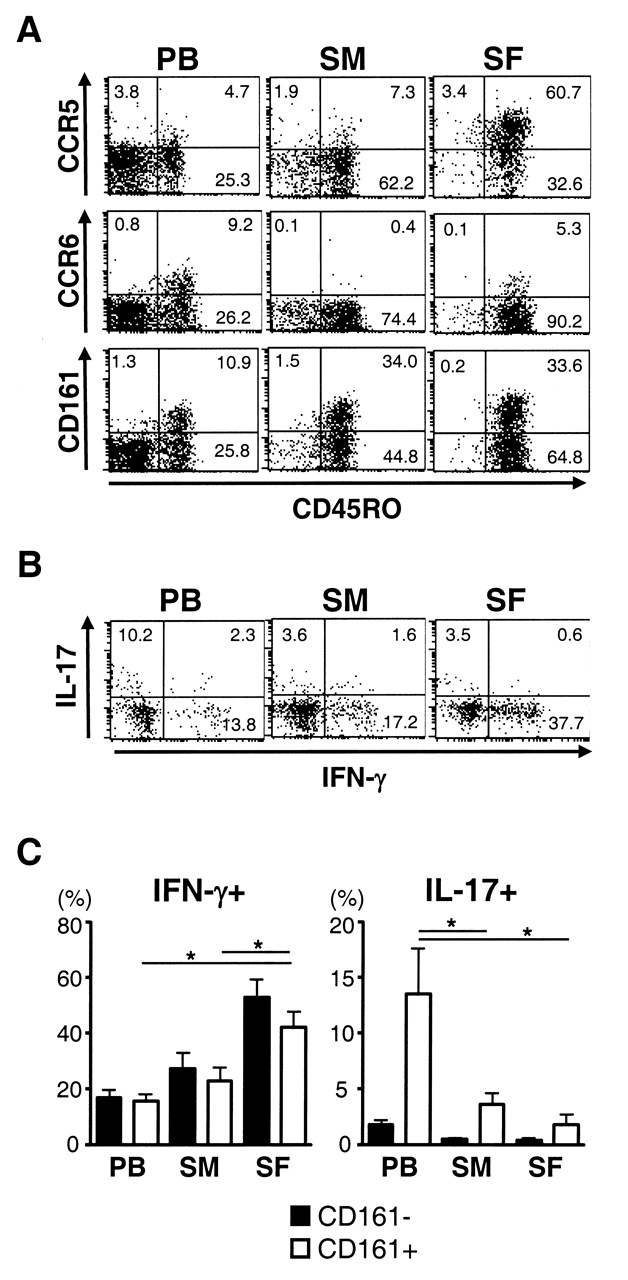
Expression of Th17-associated molecules on CD4 T cells in RA. (A) Expression of surface molecules on peripheral blood (PB), synovial membrane (SM), and synovial fluid (SF) cells of RA patients was analyzed. Representative dot plots after gating on CD4+ cells are shown. (B) Intracellular staining of cytokines was performed after brief stimulation of mononuclear cells with PMA and ionomycin. Representative dot plots after gating on CD161+CD4+ cells are shown. (C) Mean percentage of IFN-γ+ (left) and IL-17+ cells (right) in CD161– and CD161+ CD4+ cells in 5 RA patients is shown. *p < 0.05.

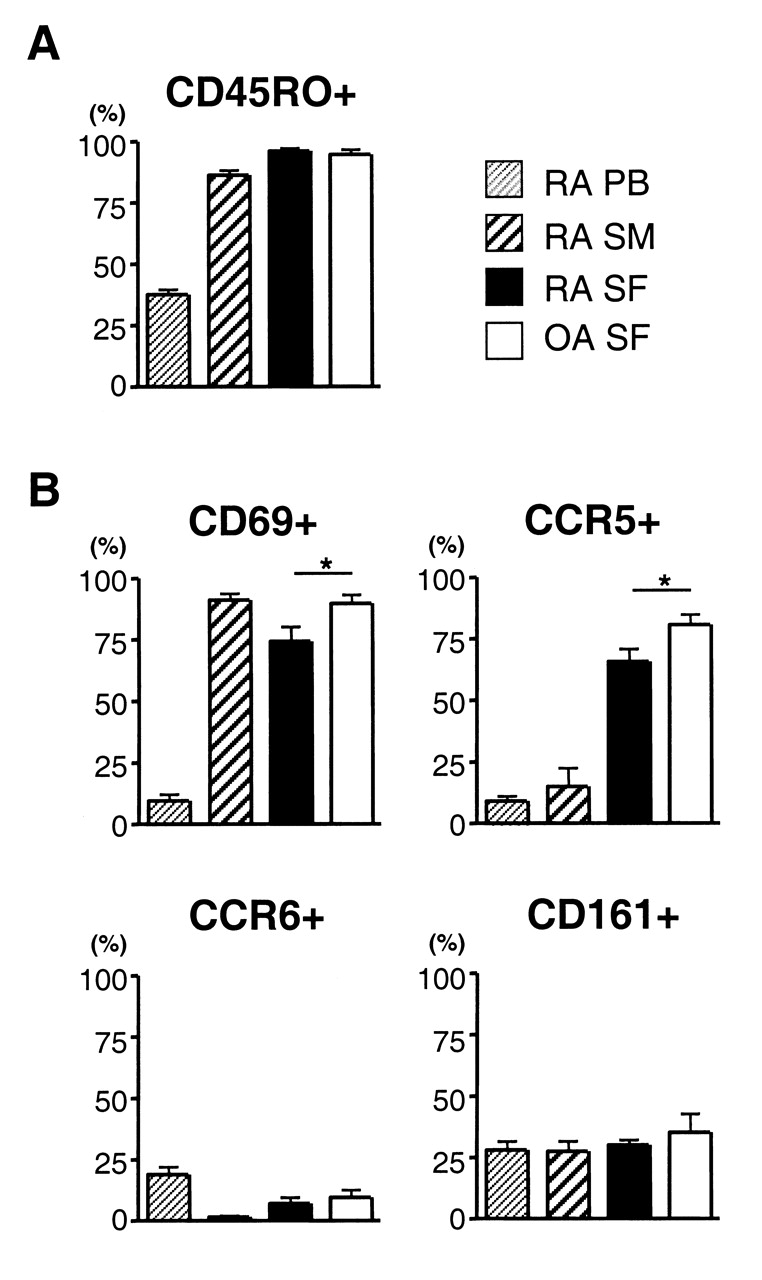
Expression levels of surface molecules on RA and OA CD4 T cells. (A) Mean percentages of CD45RO+ cells in CD4+ cells and (B) mean percentages of CD69+, CCR5+, CCR6+, and CD161+ cells in CD45RO+CD4+ cells in peripheral blood (PB), synovial membrane (SM), and synovial fluid (SF) of RA (n = 9) and SF of OA (n = 8) are shown. *p < 0.05, between RA and OA SF.
CD4 T cells in RA and OA joints were quantitatively but not qualitatively different
Although increased IL-17 production in RA joints was observed by comparison with OA joints, this could be due to the different numbers of CD4 T cells in the joints. We confirmed that there was a large difference in the absolute number of CD4 T cells between RA and OA SF (mean ± SD: RA, 2.5 × 105 ± 1.7 × 105/ml; OA, 6.6 × 103 ± 10.8 × 103/ml; p < 0.05). This also indicated that the cohort of RA patients in the study had joint effusion resulting from active RA inflammation, not from secondary OA. Indeed, serum C-reactive protein values of patients with RA were above the detection limit (Table 1). We also noted that the proportion of CD4+ cells in T cells (CD3+ lymphocytes) in the joint was also higher in RA than in OA, although it was slightly lower than the proportion in PB even in RA (data not shown).
We then investigated whether there are qualitative differences between CD4 T cells in RA and OA joints. CD4 T cells in OA joints were highly enriched for CD45RO+ cells, similar to CD4 T cells in RA joints (Figures 3A, 4A). The expression of HLA-DR and CD69 was upregulated on CD45RO+ CD4 T cells in the joints. The expression of CD69 on CD45RO+ CD4 T cells was even higher in OA than in RA (Figure 3B). Upregulated expression of CCR5 in SF CD4 T cells was also observed in OA, which was again higher than that in RA. CD161+ cells lacking CCR6 expression were abundant. There was no difference in the expression levels of these molecules between RA and OA SF CD4 T cells (Figure 3B). Although we were not able to statistically analyze the phenotype of CD4 T cells in OA SM due to the small number of samples, it was similar to those in RA SM. CD4 T cells in OA joints also lacked CD28-negative CD57+ cells. Further, phenotypes of CD8 T cells in OA joints resembled those in RA joints (Figure 4B). Thus, there seemed to be only a quantitative, not a qualitative difference in T cells infiltrated into RA and OA joints.
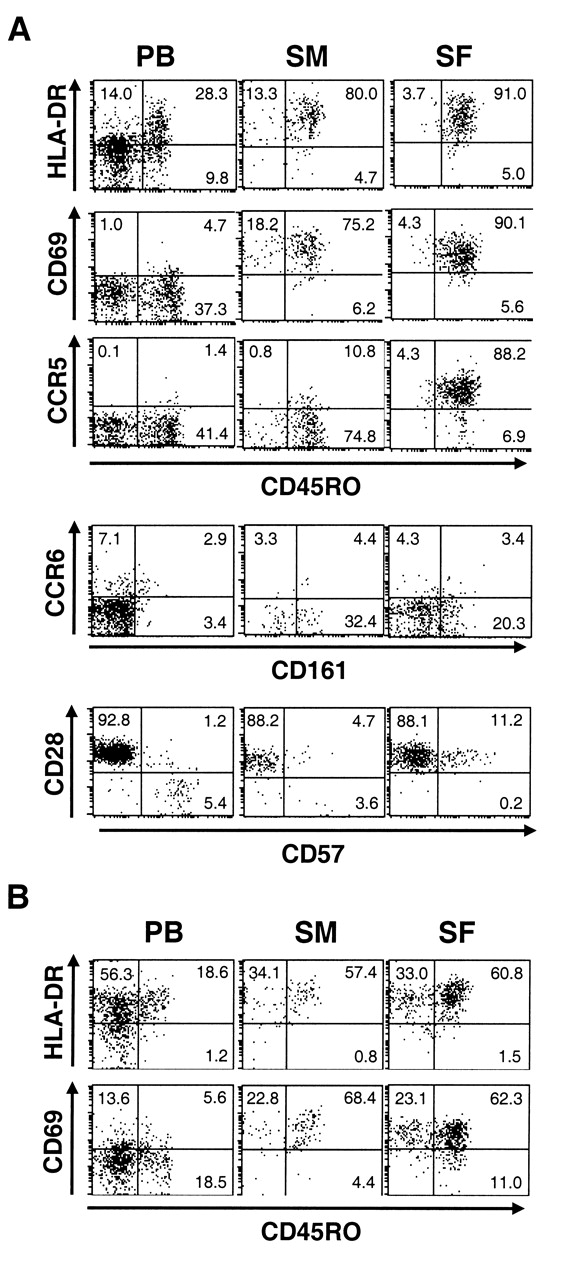
Expression of surface molecules on T cells in OA. Mononuclear cells isolated from peripheral blood (PB), synovial membrane (SM), and synovial fluid (SF) of OA patients were analyzed for the expression of surface molecules. Representative dot plots after gating on (A) CD4+ or (B) CD4–CD3+ cells are shown.
Th1 cells predominate in both RA and OA joints
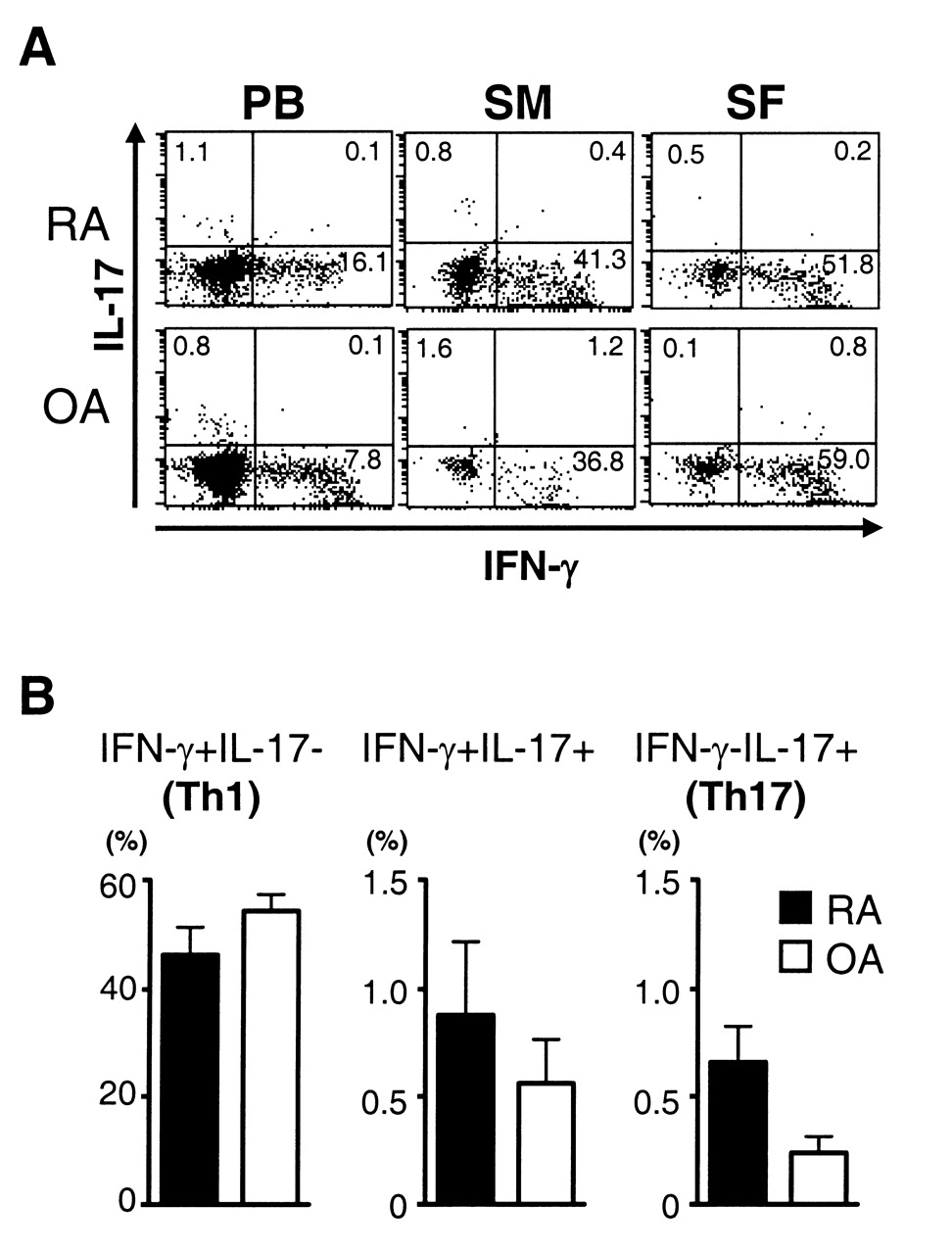
We then compared the profiles of cytokine production between RA and OA CD4 T cells (Figure 5A). Similar to CD4 T cells in RA joints, a large portion of CD4 T cells in OA joints were Th1 cells producing only IFN-γ, whereas either Th17 cells or IL-17/IFN-γ double-producers were scarcely found. Most Th1 cells in OA and RA joints were capable of producing tumor necrosis factor-α (data not shown). There was statistically no significant difference in the frequency of Th1 cells between RA and OA SF (Figure 5B). Thus, the predominance of Th1 cells is not a unique feature of CD4 T cells infiltrated into RA joints. The frequency of Th17 cells tended to be lower in OA SF than in RA SF, although the difference was not significant (p = 0.10).

Cytokine production of RA and OA CD4 T cells. (A) Intracellular staining of cytokines was performed after brief stimulation of the mononuclear cells with PMA and ionomycin. Representative dot plots after gating on CD4+ cells are shown. (B) Mean percentages of IFN-γ+IL-17– (Th1, left), IFN-γ+IL-17+ (middle), IFN-γ–IL-17+ (Th17, right) cells in CD4+ cells in RA and OA SF are shown. Statistically significant difference was not detected in all these populations between RA and OA (p > 0.05).
DISCUSSION
Despite the prevailing belief that RA is a Th17-mediated disease, we unexpectedly found in a previous study that Th1 but not Th17 cells predominated in RA joints7. We found here that CD4 T cells expressing CCR6, a marker of Th17 cells, were not enriched in RA joints, although the importance of CCR6 in T cell accumulation in inflamed joints has been described in mice18. Consistent with our results, other reports showed no increase of CCR6+ CD4 T cells in RA joints19,20. Although CD161 was expressed on a substantial portion of CD4 T cells in any site, CD161+ CD4 T cells in the joints were enriched with Th1 cells similar to CD161-negative CD4 T cells. Therefore, the unresponsiveness of CD161+ cells seems unlikely to be the explanation for the paucity of IL-17-producing cells in RA joints. It is possible that some CCR6-negative Th1 cells in the joint, if not all, originated from Th17 cells, since it is known that Th17 cells convert to Th1 cells in the presence of IL-12, which is produced in RA joints5,21.
The abundance of Th1 cells in RA joints suggests their involvement in disease pathogenesis. However, we found that CD4 T cells in OA joints were also highly enriched for Th1 cells. Expression of CCR5, a marker of Th1 cells, was detected on the majority of CD4 T cells in both RA and OA SF. Importantly, almost all CD4 T cells in OA joints expressed activation markers at even higher levels than those in RA joints. Consistent with our results, immunohistological analysis detected activated T cells and their production of IFN-γ in OA synovium22,23,24. We found that even CD8 T cells in both RA and OA joints uniformly exhibited the activated T cell phenotype, and the percentage of CD8 T cells was higher in the joints than in PB in both RA and OA. These results indicate that the predominance of activated Th1 cells in the joints does not necessarily mean that the inflammation is driven by autoreactive Th1 cells, because OA is unlikely as an autoimmune disease. Most activated T cells found in the joints might accumulate in an antigen-nonspecific manner in response to local inflammation. In support of this hypothesis, accumulation of virus-specific memory T cells has been detected in RA joints25,26. Oligoclonality of joint-infiltrating T cells was observed not only in RA but also in OA27. Interestingly, it was demonstrated that T cells in RA joints were more like cytokine-activated T cells than antigen-activated T cells28,29. This seems especially reasonable for SF T cells, which might not come in contact with antigen-presenting cells. Such cytokine-activated T cells secrete IFN-γ28. IFN-γ was detected in RA joints, albeit at a low level23,30. Therefore, the role of Th1 cells in human RA remains an open question.
Although less likely, another possible explanation for our results is that OA is also a Th1-mediated autoimmune arthritis. Indeed, there are lines of evidence suggesting the involvement of T cells in the pathogenesis of OA31. However, it should be kept in mind that RA causes erosive changes, while OA conversely induces bone formation, indicating the presence of qualitative differences in the inflammation. A more challenging possibility is that even RA inflammation is not driven by autoreactive CD4 T cells. In this scenario, autoantibody production might be a consequence of chronic stimulation of immune systems, and T cells could be involved in the pathogenesis totally in a “bystander” role. This hypothesis requires appropriate explanation for such chronic and aggressive synovitis, but it is missing. On the other hand, the self-reactivity of the joint-infiltrating oligoclonal T cells remains a fundamental question of RA.
We found no significant difference in the frequency of Th17 cells between OA and RA joints, although the frequency tended to be lower in OA than in RA. However, given the large number of total CD4 T cells in RA joints, there are considerable numbers of Th17 cells. Indeed, anti-IL-17 therapy seems to have some beneficial effect on RA32. Th17 cells in RA joints could be either etiologic autoreactive T cells that expanded locally or bystander T cells that migrated toward CCL20 in the joints18. However, the frequency of Th17 cells did not increase in the joints compared with PB. Histological analysis of RA SM also showed very low frequency of T cells producing IL-1733. In this regard, it was recently reported that most IL-17-positive cells in RA SM comprised mast cells but not T cells34, which provides an alternative explanation for the elevated IL-17 levels in RA joints.
One of the drawbacks of our study is the relatively long duration of disease. Many patients showed an advanced stage of joint damage as a consequence. Various therapeutic interventions might also affect the functions and activity of T cells. In this regard, it is notable that a recent study examining DMARD-naive patients with early RA showed an increased frequency of PB Th17 cells35. The level of IL-17 in SF was increased only in early RA36. Nevertheless, we believe our results are valuable from a clinical viewpoint. Since the goal of RA therapy is to halt chronic inflammation, it is important to elucidate the mechanism of chronic inflammation. Notably, T cell-targeting therapies such as CTLA4-Ig show clear effects even in established RA4. In addition, the erosive changes of the bone, another hallmark of RA, progress even in patients with long disease duration. Thus further investigations on chronic T cell responses in RA joints are merited.
Footnotes
-
Supported in part by the program of Founding Research Centers for Emerging and Reemerging Infectious Disease commissioned by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
- Accepted for publication March 7, 2011.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}