

Abstract
Objective. Tumor necrosis factor-α (TNF-α) is a multifunctional proinflammatory cytokine that influences the pathogenesis of Takayasu arteritis (TA). There is still no evidence of the relationship between TNF-α gene promoter polymorphisms and TA. We examined whether variations in the TNF-α promoter region may lead to TA susceptibility and disease progression.
Methods. Five TNF-α gene promoter polymorphisms (−238G/A, −308G/A, −857C/T, −863C/A, and −1031C/T) were analyzed in 110 Chinese Han patients with TA, with a control group of 362 unrelated healthy individuals. Genotypes of TNF-α gene promoter polymorphisms were identified by direct sequencing. TNF-α plasma concentrations were determined by ELISA.
Results. Our results indicated that the frequency of the −863A allele was significantly lower in the patients with TA than in the controls (18.2% vs 25.7%; p = 0.011), but the significance was lost after Bonferroni correction (pc = 0.055). The frequency of −863CA/AA genotypes was significantly lower in the patients with refractory TA than in those with the 863CC genotype (22.4% vs 44.2%; pc < 0.01). The frequency of the GGCCT haplotype was significantly higher in patients than in the controls, while the frequencies of GGCAT and GGCCC haplotypes were significantly lower in patients than in controls. The plasma TNF-α concentrations were significantly lower in the subjects carrying the −863A allele than in those without. Patients with active TA had a significant increase in plasma levels of TNF-α compared with remission patients and the control group.
Conclusion. Polymorphisms of the TNF-α promoter are not associated with TA in the Chinese Han population. The A allele of the −863C/A polymorphism is associated with decreased TNF-α expression, which might affect medical treatment.
Takayasu arteritis (TA) is a chronic large-vessel vasculitis that mainly affects the aorta and its major branches. TA typically presents before the age of 40 years. Vessel wall inflammation has been suspected of causing localized stenosis, vascular occlusion, dilatation, and aneurysm formation1,2. The pathogenesis of TA is still unclear, but it is widely believed to be a genetic-related autoimmune disease. The aortic inflammatory changes are associated with activated dendritic cells, αß and γδ T lymphocytes, B lymphocytes, macrophages, and multinucleated giant cells3,4. At the same time, the production of inflammatory cytokines is markedly increased5,6,7. Therefore, cellular and humoral immunity play major roles in the physiopathology of TA. The HLA gene has been found to be associated with TA susceptibility in different races and ethnicities8,9. Our previous study has indicated that the HLA-DRB1*04, DRB1*07, DPB1*09, and DPB1*1701 alleles may be genetic susceptibility factors for TA in the Chinese population10,11.
Tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine released from mononuclear cells, natural killer cells, and T cells. It amplifies the inflammatory response and upregulates MHC antigen expression on vascular cells. Studies have shown that TNF-α plays pivotal roles in immune system dysfunction and the development of blood vessel damage in TA8,12,13. Therefore, TNF-α is an essential cytokine in the pathogenesis of all kinds of inflammations, especially autoimmune diseases such as rheumatoid arthritis (RA) and multiple sclerosis (MS). Recent studies have demonstrated that serum TNF-α levels are elevated in patients with TA, especially in the active cases14,15. The constitutive messenger RNA expression of TNF-α from peripheral blood mononuclear cells was significantly higher in patients with TA16. Anti-TNF-α therapy has been used in some patients with TA who were undergoing refractory or standard immunosuppressive treatments. Dramatic improvement has been observed after such intervention17,18,19. Hence, TNF-α might be a key mediator in the development of TA, and the inflammatory processes in TA might be primarily regulated by TNF-α.
The TNF-α gene locus is localized within the class III region of the human MHC on chromosome 6p21.31. The variations in TNF-α gene, especially in the promoter, have been proven able to influence TNF-α expression and production. Previous studies have shown that TNF-α production is influenced by polymorphisms in the TNF-α gene, categorizing individuals as high or low TNF-α producers20. The genetic variations in the TNF-α promoter have been found to greatly influence the outcome of inflammatory and autoimmune diseases21. However, the role of TNF-α gene promoter genetic variation in the development of TA has not been investigated.
Therefore, we hypothesize that the variations in the TNF-α promoter region may confer TA susceptibility and progression of the disease.
MATERIALS AND METHODS
Subjects
A total of 110 unrelated Chinese Han patients with TA (85 women and 25 men; average age 33.7 ± 12.2 yrs) from the Fuwai Hospital were studied. All patients fulfilled the 1990 American College of Rheumatology classification criteria for TA22. A total of 362 unrelated healthy individuals served as the controls (177 women and 185 men; average age 35.8 ± 9.8 yrs). Informed consent was obtained from all individuals, and the study protocol was approved by the Ethics Committee of the Fuwai Hospital.
Clinically active TA was diagnosed based on at least 2 of the following disease features: (1) systemic signs or symptoms not attributable to other clinical conditions; (2) elevated erythrocyte sedimentation rate and C-reactive protein in the absence of infection or malignancy; (3) onset of signs or symptoms of vascular insufficiency; and (4) new vascular lesions in previously unaffected blood vessels detected on imaging through angiography, computed tomography, vascular Doppler imaging, and vascular magnetic resonance imaging23. According to this definition, 47 patients were in the active phase of disease and 63 were in the remission phase at the outset of the study. Arteriographic classification was as follows: type I, involvement of the aortic arc and its main branches; type II, involvement of the thoracic and abdominal aorta; type III, involvement of the whole aorta; and type IV, involvement of the pulmonary artery24,25.
Clinically difficult to treat cases were defined using the following criteria: patients who required toxic doses of corticosteroids and/or conventional immunosuppressive agents to maintain remission, or who had experienced multiple relapses after standard treatments.
Determination of plasma concentrations of TNF-α
Plasma concentrations of TNF-α were determined in 75 patients and 45 controls who were randomly selected. The patients were divided into active and remission groups according to the above criteria. To minimize the potential confounding effects, patients in remission were selected only when they had a stabilized disease for more than 6 months. Venous blood was collected into a whole-blood tube with K3EDTA and centrifuged at 2000 ×g for 10 min at 4°C. Plasma samples were then frozen and stored at −80°C until the assays were performed. TNF-α plasma levels were determined by ELISA with commercial kits (Rapidbio, West Hills, CA, USA) according to the manufacturer’s instructions.
Genotyping of the polymorphism of TNF-α gene promoter
Genomic DNA was extracted from peripheral blood cells by a common salting-out procedure and dissolved in Tris-HCl buffer (10 mmol/l, pH 8.0) containing 1 mmol/l EDTA, and stored at −80°C until use. The TNF-α polymorphism data were collected from a public database (US National Institutes of Health; http://www.ncbi.nlm.nih.gov/). Only single-nucleotide polymorphisms (SNP) with minor allelic frequencies above 5% in the literature or with previous epidemiologic findings, which indicate associations with autoimmune diseases, were chosen. A minimum r2 of 0.8 was set as the threshold for all analyses. Five polymorphisms of the TNF-α promoter were selected, namely −238G/A (rs361525), −308G/A (rs1800629), −857C/T (rs1799724), −863C/A (rs1800630), and −1031C/T (rs1799964). The primers 5′-TTC AGG GAT ATG TGA TGG AC-3′ and 5′-ATA AAG GCA GTT GTT GG CA-3′ were used to amplify a 1100 bp DNA fragment containing the −7 to −1107 loci of the TNF-α promoter. Polymerase chain reaction (PCR) was performed in a 25 μl reaction mixture containing 1 U DNA Taq polymerase (Shanghai Biocolor, Shanghai, China), 50 ng genomic DNA, 1 × buffer, 1.5 mmol/l of MgCl2, 0.2 mmol/l primers, and 0.08 mmol/l deoxyribonucleoside triphosphate. PCR cycling conditions were as follows: DNA denaturation for 2 min at 94°C, followed by 35 cycles of amplification (30 s at 94°C, 30 s at 64°C, and 30 s at 72°C), and a final extension step at 72°C for 5 min. The polymorphisms of TNF-α promoter were identified by direct sequencing of PCR products with a Biosystems 3700 capillary sequencer.
Statistical analysis
Continuous variables with a normal distribution were expressed as mean ± SD. The differences in quantitative variables among groups were analyzed using the independent Student’s t test. The distribution of TNF-α plasma levels was skewed (non-normal), so the Kruskal-Wallis test was used to compare the plasma TNF-α levels among different groups. The chi-squared analysis and Fisher’s exact tests were used to test for qualitative variables, genotypic/allelic frequencies, and the Hardy-Weinberg equilibrium (HWE) of the variants. The associations between the variants and TA were determined using unconditional logistic regression models. The covariates for the logistic regression models included conventional factors such as age and sex. The corrected p values were adjusted by applying Bonferroni correction. Haplotype frequencies of the TNF-α gene promoter SNP were determined with SHEsis online (http://analysis.bio-x.cn)26. P < 0.05 was the level of statistical significance (2-tailed). Statistical analysis was performed using the SPSS software package (version 13.0).
RESULTS
Characteristics of patients with TA
The average age at clinical disease onset was 25.47 ± 9.43 years (range 7 to 53 yrs), while the average age at diagnosis was 33.53 ± 12.14 years (range 13 to 64 yrs). There were 47 patients in the active phase of the disease and 63 in the remission phase at the outset of the study. Of the 58 cases classified as refractory, 55.4% had type III angiographic classification, while 26.8%, 17.9%, and 11.8% had types I, II, and IV, respectively.
Polymorphisms of TNF-α gene promoter associated with TA
The genotypic distributions and allelic frequencies of the 5 TNF-α gene promoter SNP are presented in Table 1. The genotype distributions in both patients and controls were all within the HWE. After using unconditional logistic regression analysis to adjust for age and sex between the 2 groups, the frequency of the −863A allele was significantly lower in the patients with TA than in the controls (18.2% vs 25.7%; p = 0.011), but the significance disappeared after Bonferroni correction (pc = 0.055). No significant associations were observed between TA and variants −238G/A, −308G/A, −857C/T, and −1031C/T.
Genotype distributions and allele frequencies of the tumor necrosis factor-α gene promoter in the study subjects. Genotype p values were obtained with multivariate unconditional logistic regression analysis by adjusting for age and sex. Genotype analysis were conducted for the dominant model (CC compared with CA + AA). p values were adjusted by Bonferroni correction.
Haplotype analysis
The haplotype frequencies are presented in Table 2. Seven major haplotypes with frequencies > 0.03 were identified. No linkage disequilibrium was found among the 5 selected SNP by SHEsis software analysis26. Haplotypes with frequencies < 0.03 in both the patients and controls are not listed in Table 2. The GGCCT (haplotype 1) of −238G/A, −308G/A, −857C/T, −863C/A, and −1031T/C polymorphisms was the most common. The frequency of GGCCT was significantly higher in the patients than in the controls (62.3% vs 51.0%; pc < 0.01). However, the frequencies of GGCAT (haplotype 3) and GGCCC (haplotype 5) were significantly lower in the patients than in the controls (haplotype 3, 0% vs 9.6%, pc < 0.01; haplotype 5, 0.5% vs 5.1%, pc = 0.017). There were no significant differences in the frequencies of 4 other major haplotypes between patients with TA and controls.
Haplotype frequencies of TNF-α gene promoter polymorphisms, arranged as −238G/A, −308G/A, −857C/T, −863C/A, and −1031T/C. Haplotypes with frequencies < 0.03 in both controls and patients were not included. p values were adjusted by Bonferroni correction.
Relationships between −863C/A polymorphism and plasma TNF-α concentrations
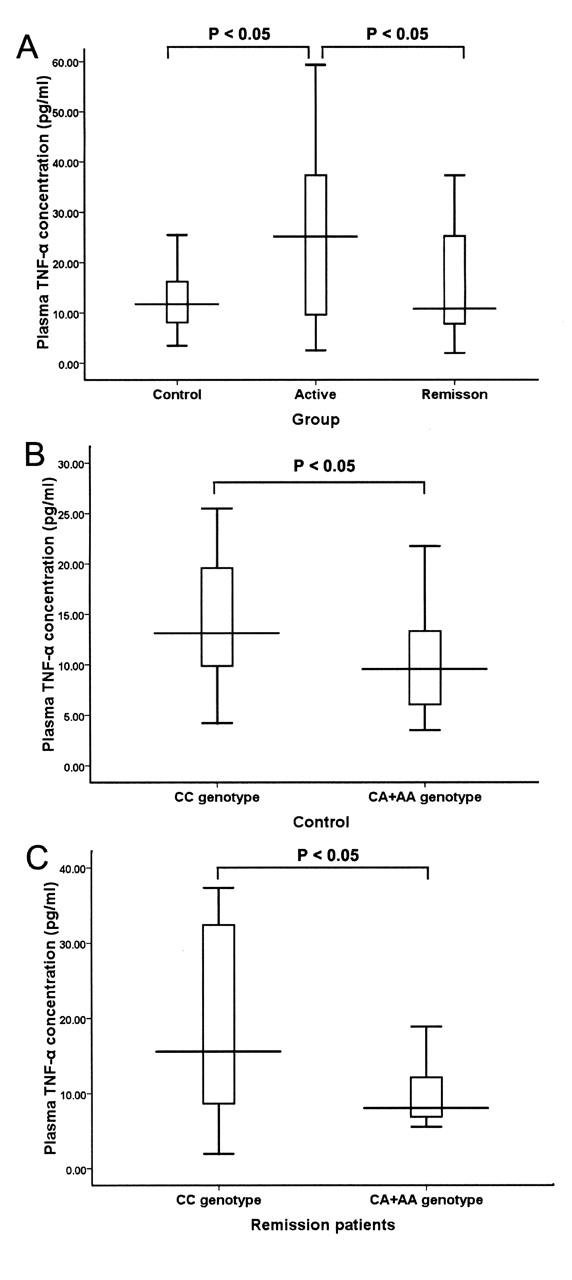
Plasma concentrations of TNF-α were determined in 75 patients and 45 controls. The patients with active disease (n = 30, 25.17 pg/ml, 25th–75th percentiles 9.54–38.36 pg/ml) had significantly increased TNF-α plasma levels compared with the patients in remission (n = 45, 10.79 pg/ml, 25th–75th percentiles 7.65–27.78 pg/ml) and controls (n = 45, 11.74 pg/ml, 25th–75th percentiles 8.04–16.48 pg/ml; p < 0.05, Kruskal-Wallis test; Figure 1A). The relationships between the −863C/A polymorphism and plasma TNF-α concentrations were analyzed among patients in remission and controls. The plasma TNF-α concentrations were significantly lower in the subjects carrying the −863A allele (n = 15, 9.55 pg/ml, 25th–75th percentiles 5.67–13.59 pg/ml) than in those without the A allele (n = 30, 13.13 pg/ml, 25th–75th percentiles 9.43–19.66 pg/ml) among the healthy controls (p = 0.037, Kruskal-Wallis test; Figure 1B). Among the patients in remission, the plasma TNF-α concentrations were also lower in the subjects carrying the −863A allele (n = 15, 8.05 pg/ml, 25th–75th percentiles 6.89–14.19 pg/ml) than in those without the A allele (n = 30, 15.59 pg/ml, 25th–75th percentiles 8.63–32.59 pg/ml; p = 0.044, Kruskal-Wallis test; Figure 1C). No significant associations were observed between the plasma TNF-α concentrations and variants −238G/A, −308G/A, −857C/T, and −1031C/T.

A. Plasma concentrations of TNF-α in patients with active Takayasu arteritis (TA) and patients in remission, compared with controls. TNF-α plasma levels are presented as boxes (25th percentile, median, and 75th percentile) and whiskers (10th–90th percentiles). B. Plasma TNF-α concentrations among the controls, with different −863C/A genotypes (the CA/AA genotype compared with the CC genotype). C. Plasma TNF-α concentrations among patients in remission, with different −863C/A genotypes (the CA/AA genotype compared with the CC genotype).
Association between polymorphisms of TNF-α gene promoter and patients with refractory TA
The association between TNF-α promoter −863C/A polymorphisms and patients with refractory TA is presented in Table 3. Genotypic analyses were conducted with the dominant model by multivariate unconditional logistic regression analysis to adjust for age and sex. The frequency of −863CA/AA genotypes was significantly lower in the patients with refractory TA than in controls (22.4% vs 44.2%, respectively; pc < 0.01). In addition, the frequency of −863CA/AA genotypes was significantly lower in the patients with refractory TA than in patients with nonrefractory TA (22.4% vs 40.3%; p < 0.05). There were no significant differences in the other 4 variants between patients with refractory TA and patients with nonrefractory TA.
Association between genotype distributions of −863C/A polymorphism with patients with refractory Takayasu arteritis (TA). Genotype p value was obtained with multivariate unconditional logistic regression analysis adjusting for age and sex. Corrected p value was adjusted by Bonferroni correction.
DISCUSSION
TA is characterized by granulomatous inflammation of the aorta and its major branches. It is clinically characterized by exacerbations and remissions. A previous study revealed that serum TNF-α levels were significantly higher in patients with active TA15. Moreover, Tripathy, et al observed increased constitutional TNF-α expression in the peripheral blood mononuclear cells of 10 patients with TA16. We also observed the significant increase of TNF-α level in patients with active TA. The results suggest that TNF-α is a useful marker for monitoring the activity of TA, similar to endothelin 1 and circulating endothelial cells27. However, the role of genetic variations of the TNF-α gene promoter in TA susceptibility has not been investigated. In this case-control study, 5 polymorphisms were identified in the TNF-α promoter region by genomic sequencing. Although the frequency of −863CA/AA genotypes in patients with TA was lower compared with that of the control group, the significance of −863A/C susceptibility disappeared after Bonferroni correction was applied strictly. But the frequency of the −863CA/AA genotypes was significantly lower among the patients with refractory TA. Plasma TNF-α concentrations were significantly lower among subjects carrying the −863A allele than in those without. So our study shows that the A allele of −863C/A polymorphism is associated with reduced TNF-α serum levels and therapeutic response among the 5 variants (−238G/A, −308G/A, −857C/T, −863C/A, and −1031C/T).
The level of TNF-α production in healthy individuals shows wide and stable variation, with high and low producer phenotypes present in the population20. Previous studies have shown that TNF-α expression is influenced by functional polymorphisms in their gene loci, which may contribute to the susceptibility or severity of the autoimmune and inflammatory diseases, such as RA, MS, ankylosing spondylitis, and asthma21. In our study, results show that the frequency of the −863A allele is significantly lower among patients with refractory TA. Further, carriers of the −863A allele had significantly lower TNF-α levels than carriers of −863CC, suggesting that this polymorphism might affect TNF-α expression at the transcriptional level, and contribute to the outcome of TA. Indeed, Skoog, et al found that the rare −863A allele is associated with 31% lower transcriptional activity in chloramphenicol acetyltransferase reporter gene studies in human hepatoblastoma cells. Moreover, the 10 bp sequence (GGG GAC CCCC), which carries the −863C/A polymorphism, shows considerable similarity with the consensus sequence for the nuclear factor-κB (NF-κB) binding site28, and alters the relative binding affinities of different forms of the NF-κB complex, resulting in different transcriptional activities29. Therefore, the −863A allele might have a protective role in the progress of TA by inhibiting NF-κB binding, which leads to decreased TNF-α transcription and reduced TNF-α level in patients with TA.
TNF-α-producing CD3+ T cells are higher in active TA14. In our study, the patients with active disease experienced significant increases in TNF-α plasma level compared with the patients in remission. The results confirm that TNF-α plays a key role in the pathophysiology of TA. The traditional medical treatments for patients with active TA are immunosuppressive agents, including corticosteroids and methotrexate. However, patients with refractory disease require toxic doses of corticosteroids or concomitant treatment with other immunosuppressive drugs to achieve disease remission. In our patients, the frequency of the −863A allele was significantly lower than in the controls or in patients with nonrefractory disease, a finding that suggests that the genetic variants of the TNF-α promoter might affect the severity of inflammatory reactions by regulating TNF-α production. Clinically, carriers of the −863A allele with lower TNF-α production might respond better to medical treatment. Anti-TNF-α agents have been proven effective in the treatment of patients with TA, especially in refractory cases17,18,19,30. High TNF-α production might be associated with better responses to anti-TNF-α therapy. Identifying patients with higher expression levels of TNF-α could be useful for the treatment of TA. Further studies are therefore required.
The haplotype analysis could provide further insight into the role of the TNF-α promoter in the molecular pathogenesis of TA. In our study, 7 major haplotypes were found. Among them, the haplotype GGCCT was the most represented in both the patient and control groups, and it was associated with increased risk of TA. Haplotypes GGCAT and GGCCC were more frequent in the control group compared with the patients, but both levels of frequency were relatively low in the Chinese Han population.
The limitations of our study are as follows: first, our findings, which are based on a relatively large population of patients with TA, have not been confirmed in a second independent cohort, and the results await replication in different populations. Second, in vitro experiments are required to investigate the underlying mechanism of −863C/A polymorphism in TNF-α production.
TNF-α plasma levels are elevated in the active stage of TA, which may be a key cytokine in TA development and progression. The A allele of the −863C/A polymorphism is associated with decreased TNF-α expression in the Chinese Han population, which might have implications for clinical treatment. The different TNF-α gene promoter polymorphism haplotypes might contribute to the molecular pathogenesis of TA. Therefore, further study of the underlying mechanism is warranted.
Footnotes
-
Supported by the National Natural Science Foundation of China (grant no. 30770859) and the Research Fund for the Doctoral Program of Higher Education of China (grant no. 20091106110014).
- Accepted for publication July 25, 2011.








{kind=link}